
Theoretical Study of the Kinetic-Thermodynamic Competition between the σ-Complexes Obtained by the Reaction of the Methoxy Ion on the 7- Methyl 4-Nitro Benzofuroxan
Download this article as:
ABSTRACT:Superelectrophilic compounds present interesting structural properties, chemical and biological behavior. A great number of most studied compounds include benzofuroxanic ring and differ by the substitution mode. DFT/B3LYP study have been performed in order to interpret the kinetic-thermodynamic competition between the σ-complexes obtained by the reaction of the methoxy ion on the 7- methyl 4-nitro benzofuroxane. Geometry, atomic charge distribution, thermodynamic parameters have been calculated for all possible products. The most unexpected result is to find that the most stable compound does not have been detected experimentally
KEYWORDS:Anticancer; Nitrobenzofuroxan (NBF); Mutagenicity; Superelectrophilic Compounds
Introduction
The high capacity of nitrobenzofuroxans to undergo covalent addition or substitution processes is at the origin of a great interest in the last decades. This research is partly driven by their pharmacological properties and the importance of these species for biological processes.1-7 In particular, nitrobenzofuroxans were synthesized for testing as potential anti-rheumatic drugs and were also tested for their role in the mutagenicity in Salmonella typhimurium strains TA98 and TA100.8 Nitrobenzofuroxans were also tested as anti cancer in Salmonella typhimurium. Their mutagenicity was not found to be correlated to the previously established anti leukemic activity9. Those compounds were also strongly investigated from chemical activity point of view.10-19
The problem that we deal with concerns the difference of behavior between nitro and dinitro benzofuroxans (DNBF). Placed in the same experimental conditions, the two compounds exhibit different chemical reactivity20 as it is shown in Fig. -1. The action of the methoxy ion leads to the anion DNBF– 4’ by means of slow chemical ionization process on the hydrogen of the 7-methyl. Any other compound has been observed. This reaction put in evidence an expected superelectrophilic property which has surprised by its very low pKa equal to 2.4. Such value places this compound very close mineral acids21. This acidity concerns proton of the 7-methyl and can be interpreted by the great stability of its charged form, DNBF–. However, NBF leads in presence of the methoxy ion at σ-complexation reactions. Two products, 2 and 3 have been detected exhibiting a kinetic/thermodynamic competition. Indeed, as the reaction progresses, the formed quantity of 2 diminishes in favor of the 3. We present in Fig. -1, a hypothetical reaction schema of all the possible compounds witch may exist similarly for NBF and DNBF in presence of methoxy anion but we will dual only with NBF in order to interpret its behavior. The study of DNBF has been accomplished previously.22
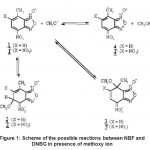 |
Figure 1: Scheme of the possible reactions between NBF and DNBG in presence of methoxy ion |
Results and Discussion
We have optimized the geometry of all the studied compounds using the package- program Gaussian 03 by means of the DFT/B3LYP technique23. Gaussian type basis sets (4s4p1d/ 3s3p1d) were employed for carbon; nitrogen and oxygen including pseudopotentials of Barthelat and Durand24,25. We checked that all the obtained structures concern minimum energy values on the potential surface for the compounds 1 to 4. Our obtained values close to the experimental ones10 determined by x-ray diffraction.
Because several electronic effects intervene simultaneously, it is difficult to propose a semi-empirical formulation of NBF. We show tentatively in Fig. – 2 a mechanism driving with the formation of the furaxonyl ring from two nitroso functions. We see that the two electrons of double bond C3-C8 are employed to form the two C==N bonds. This situation seems handicapping the communication between the electronic parts of benzyl and the furaxonyl rings.
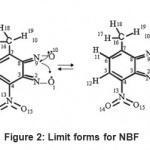 |
Figure 2: Limit forms for NBF |
We can consider that the complexation reaction between NBF and the ion methoxy as a charge controlled one. As negative specie, the latter plays the role of electrophilic compound but the former is a nucleophilic one. Thus, it is necessary to start with a systematic analysis of the atomic charge distribution of NBF. Data of table 1 gathers atomic Mulliken charge of all the studied compounds 1 to 4. According to a control charge process, the attack of the ion methoxy can be only in three centres: C3, C5 and C7 because of their positive charges. The complexation on C3 was never observed, certainly because of steric effects, thus the nucleophilic attack is envisaged only on C5 and C7. The charge of H17 on benzyl ring is more positive then the hydrogen atoms H11 , H12 one and diminishes the probability of a possible attack in the latter centres.
Table 1: Atomic Charge Q of Compounds 2, 3, 4 and their variations calculated relatively to compound 1.
| Compund | 1 | 2 | Q(2,1) | 3 | Q(3,1) | 4 | Q(4,1) |
| O1 | -0.18 | -0.22 | -0.04 | -0.19 | -0.01 | -0.24 | -0,06 |
| N2 | 0.03 | -0.01 | -0.04 | -0.05 | -0.08 | -0.04 | -0,07 |
| C3 | 0.01 | -0.01 | 0.00 | 0.02 | 0.03 | 0.00 | 0,01 |
| C4 | -0.03 | 0.03 | -0.02 | 0.00 | -0.05 | -0.04 | -0,09 |
| C5 | 0.05 | -0.05 | -0.02 | -0.08 | -0.04 | -0.05 | -0,02 |
| C6 | -0.13 | -0.14 | -0.01 | -0.20 | -0.07 | -0.17 | -0,04 |
| C7 | 0.04 | 0.01 | -0.03 | 0.08 | 0.04 | 0.08 | 0,04 |
| C8 | -0.16 | -0.15 | 0.01 | -0.14 | 0.02 | -0.19 | -0,02 |
| N9 | 0.37 | 0.37 | 0.00 | 0.42 | 0.05 | 0.38 | 0,01 |
| O10 | -0.22 | -0.30 | -0.08 | -0.34 | -0.12 | -0.32 | -0,10 |
| H11 | 0.15 | 0.12 | -0.03 | 0.11 | -0.04 | 0.06 | -0,10 |
| H12 | 0.15 | 0.12 | -0.03 | 0.12 | -0.04 | 0.10 | -0,05 |
| N13 | 0.40 | 0.39 | -0.01 | 0.40 | 0.00 | 0.40 | -0,01 |
| O14 | -0.33 | -0.35 | -0.02 | -0.45 | -0.12 | -0.45 | -0,12 |
| O15 | -0.29 | -0.35 | -0.06 | -0.41 | -0.12 | -0.41 | -0,12 |
| C16 | -0.25 | -0.24 | 0.01 | -0.26 | -0.01 | -0.27 | -0,02 |
| H17 | 0.13 | 0.12 | -0.01 | 0.09 | -0.03 | 0.06 | -0,07 |
| H18 | 0.13 | 0.14 | 0.01 | 0.07 | -0.06 | 0.10 | -0,02 |
| H19 | 0.16 | 0.16 | 0.01 | 0.10 | -0.06 | **** | |
| O20 | **** | -0.60 | -0.47 | **** | |||
| C21 | **** | 0.04 | -0.02 | **** | |||
| H22 | **** | -0.02 | 0.09 | **** | |||
| H23 | **** | -0.02 | 0.09 | **** | |||
| H24 | **** | -0.03 | 0.04 | **** |
There are various parts of the benzyl rings and/or of furoxanyl which are candidate with the conjugation. To specify such phenomena requires the localization of charge transfer and the determination of the bond indices. From bond lengths shown on table 2, we notice that the bond orders change clearly from a compound to the other. A fast outline of these values enables us to affirm that the two benzyl and furoxanyl rings are coplanar as it intuitively forecasted. Some bond indices are easily allotted because they formally relate to simple or double bonds. However, many lengths of bonds are intermediate between these two limits indicating that the presence of the conjugation in several part of these molecules. In order to quantify the electronic transfer in intermediary bonds, NBO calculations26 were performed in order to determine the Wiberg indices (Iw). The obtained values are shown in table 2. In general we can attribute a single character at a bond if Iw is smaller to 1 and we can consider bond as double for if Iw is greater than 1.2.
Table 2: Bond Lengths L in ú bond order i and the Wiber indice Iw for studied compounds. (S) is for simple Bond. (D) for Double and (I) for intermediary.
| Compound | 1 | 2 | 3 | 4 | ||||||||||||||||
| Bond | L | i | Iw | L | i | Iw | L | i | Iw | L | i | Iw | ||||||||
| C6-C7 | 1.35 | D | 1.53 | 1.33 | D | 1.51 | 1.52 | S | 1.12 | 1.47 | S | 1.12 | ||||||||
| C5-C6 | 1.44 | I | 1.2 | 1.52 | S | 1.12 | 1.34 | D | 1.53 | 1.34 | D | 1.61 | ||||||||
| C4-C5 | 1.36 | D | 1.54 | 1.50 | S | 1.06 | 1.44 | I | 1.13 | 1.44 | I | 1.21 | ||||||||
| C4-C3 | 1.45 | I | 1.13 | 1.48 | S | 1.08 | 1.43 | I | 1.14 | 1.43 | I | 1.14 | ||||||||
| C3-C8 | 1.43 | I | 1.13 | 1.48 | S | 1.09 | 1.45 | I | 1.12 | 1.45 | I | 1.12 | ||||||||
| C7-C8 | 1.44 | I | 1.17 | 1.46 | I | 1.12 | 1.52 | S | 1.17 | 1.47 | S | 1.06 | ||||||||
| C8-N9 | 1.32 | D | 1.22 | 1.29 | D | 1.33 | 1.29 | D | 1.35 | 1.29 | D | 1.32 | ||||||||
| C3-N2 | 1.29 | D | 1.51 | 1.30 | D | 1.52 | 1.29 | D | 1.52 | 1.29 | D | 1.51 | ||||||||
| N9-O1 | 1.33 | S | 0.88 | 1.33 | S | 0.94 | 1.33 | S | 1.02 | 1.34 | S | 0.87 | ||||||||
| O1-N2 | 1.37 | S | 1.09 | 1.30 | S | 1.03 | 1.39 | S | 1.06 | 1.39 | S | 1.04 | ||||||||
| N9-O10 | 1.21 | D | 1.49 | 1.25 | D | 1.45 | 1.24 | D | 1.49 | 1.24 | D | 1.40 | ||||||||
| C6-H11 | 1.08 | S | 0.98 | 1.08 | S | 0.98 | 1.08 | S | 0.98 | 1.08 | S | 0.98 | ||||||||
| C5-H12 | 1.07 | S | 0.97 | 1.08 | S | 0.97 | 1.07 | S | 0.98 | 1.07 | S | 0.97 | ||||||||
| C4-N13 | 1.45 | S | 0.94 | 1.37 | I | 1.15 | 1.38 | I | 1.17 | 1.38 | I | 1.12 | ||||||||
| N13-O14 | 1.20 | D | 1.47 | 1.23 | D | 1.36 | 1.23 | D | 1.32 | 1.23 | D | 1.35 | ||||||||
| N13-O15 | 1.19 | D | 1.50 | 1.22 | D | 1.41 | 1.22 | D | 1.42 | 1.21 | D | 1.39 | ||||||||
| C7-C16 | 1.50 | S | 1.03 | 1.51 | S | 1.07 | 1.53 | S | 1.12 | 1.34 | D | 1.67 | ||||||||
| C16-H17 | 1.09 | S | 0.99 | 1.09 | S | 0.98 | 1.09 | S | 0.97 | 1.08 | S | 0.98 | ||||||||
| C16H18 | 1.09 | S | 0.97 | 1.09 | S | 0.99 | 1.08 | S | 0.98 | 1.07 | S | 0.98 | ||||||||
| C16H19 | 1.08 | S | 0.98 | 1.09 | S | 0.97 | 1.09 | S | 0.97 | – | – | – | ||||||||
| C-O20 | – | – | – | 1.43 | S | 1.14 | 1.43 | S | 1.16 | – | – | – | ||||||||
| O20-C21 | – | – | – | 1.40 | S | 1.08 | 1.40 | S | 1.08 | – | – | – | ||||||||
| C21-H22 | – | – | – | 1.09 | S | 0.97 | 1.09 | S | 0.97 | – | – | – | ||||||||
| C21-H23 | – | – | – | 1.09 | S | 0.98 | 1.09 | S | 0.98 | – | – | – | ||||||||
| C21-H24 | – | – | – | 1.09 | S | 0.99 | 1.09 | S | 0.97 | – | – | – | ||||||||
The benzyl double bonds C==C are harshly localised between C4-C5 and C6-C7 in compounds 1, C6-C7 in complex 2, C5-C6 in compounds 3 and 4. Bonds C3-N2, C8-N9, N13-O14 and N13-O15, kept roughly their lengths which are primarily characteristics of double bond in all the studied compound. This behaviour indicates the existence of a moderated conjugation between the nitro function and the benzyl ring or between the furaxonyl ring with the benzyl one. This presence of the conjugation is confirmed by the fact that the length of the bond C4-N13 diminishes from 1.45Å in compound 1 Å to 1.37 Å in 2 to 4 which is a characteristic of bonds in mesomeric systems. Bond C6-C17 acquires a character of bond doubles only for compound 4. We present on figure 3 the more traditional manner to represent the studied molecules.
The modification observed of the localization of single or double bonds C==C generates a disturbance of the atomic distribution charge. Starting from the values collected in table 1, we can evaluate the larger modifications at -0.12 electron. The principle modifications are represented in Fig. – 3. Those modifications are moderate because the added negative charge is shared between a great number of atoms. This large distribution is at the origin of the moderate bond lengths modification in the two rings of studied molecules.
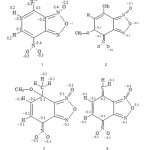 |
Figure 3: Representation 1 of the most probable structure and atomic charge variation for studied compounds 2 -4 calculated from values indicated in compound 1. |
The structural and charge distribution analysis does not show any fact which can be at the origin of the non observation of the compound 4 indeed of the great stability for its homologue 4’ and why 2 and 3 are in competition. Thus, we carry out thermodynamic calculation in order to extend our study to discuss the stability point of view. Table 3 collects variation of the enthalpy rH°, Gibbs enthalpy rG° and entropy rS° for each studied reaction. All those reactions are possible spontaneously because their rG° are negative. They are also exothermic because each rH° is negative. The values of rS° are in concord with degrees of order change. The formation of the complexes 2 and 3 increases the degree of order and generates a negative variation rS°. In contrary, the existence of the carbanion 4 kept invariable the number of particles and reasonably does not modify the entropy of the system.
Table 3: Thermodynamic parameters for studied compounds reaction of formation.
| 2 | 3 | 4 | |||||||
| H° (kcal.mol-1)
r |
-11.2 | -36.6 | -62.3 | ||||||
| G° (kcal.mol-1)
r |
-17.4 | -19 | -61.5 | ||||||
| S° (cal.mol-1.K-1)
r |
-56.5 | -58.9 | -2.8 | ||||||
The energetic diagram is described on Fig. -4. It is clear that compound 4 is the most stable. Our thermodynamic study does not give any reason for the fact that it did not be observed. The fact that the energy of formation of the complexes 2 and 3 are relatively close (The difference is only about 1.5 kcal mol-1) is in favour of the observed competition. Because compound 2 was observed in first indicates that the formation of its transition state is earlier. It must to have a barrier of formation lower than that of compound 3. A theoretical kinetic study is now carried out in order to answer those questions.
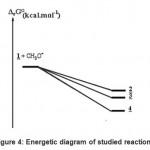 |
Figure 4: Energetic diagram of studied reactions. |
Conclusions
We studied in this paper the eventuated compound formation by the action of methoxy anion with NBF. The most important result is the moderate conjugation between the different parts of those molecules: the nitro function, the benzyl and the furaxonyl rings. The addition of the negative charge is roughly equally shared between the centres acceptors of electrons. The most unexpected result is to find that the most stable compound 4 does not have been detected experimentally. It would seem that this is due to kinetic effects consequence of the very great difference of half -time reactions between those of the formation of complexes 2 and 3 and that of the anion NBF–. Only a kinetic study will be able to give a reasonable explanation. Such work is actually performed in our laboratory.
References
- Ghosh, P. B.; Ternai, B.; Whitehouse, M. W.;J. Med Chem. , 15, 1255 (1972).
- Ghosh, P. B.; Ternai, B.; Whitehouse, M. W.; Res. Rev., 1, 159 1981.
- Terrier, F. In Nucleophilic Aromatic Displacement; Feuer, H., Ed.; VCH: New York, 1991; Chapters 1 and 2
- Crampton, M.R.S; Lunn, R.E.A.; Lucas, D, Organic and Biomolecular Chemistry, 1(19) 3438 (2003).
- Nemeikaite-Ceniene, A.;Sarlauskas, J.; Miseviciene L; Anusevicius, Z., Maroziene, A.; Cenas, N.; Acta Biochimica Polonica, 51(4), 1081 (2004).
- Asghar, B.H.M.; Crampton, M.R., Organic and Biomolecular Chemistry, 3(21), 3971 (2005).
- Eckert, F. ; Rauhut, G.; Katritzky, R.A.; Steel, P. J.; Am. Chem. Soc., 121, 6700 (1999).
- Macphee, D. G; Robert, G. P.; Ternai, B.; Ghosh P. B. ; Stephens R., Chemico-Biological Interactions, 19(1), 77 (1977).
- Thompson, S.; Kellicutt, L., Mutation Research, 48(2), 145 (1977).
- Eckert, F.,. Rauhut, G.; Katritzki A. R. ; Steel P. J.; Am. Chem. Soc. 121, 6700 (1999).
- Buncel, E.; Renfrow, R. A.; Strauss, M. J. Org.Chem., 52, 488 (1987).
- Buncel, E.; Manderville, R. A.; Dust, J. M. Chem. Soc., Perkin Trans. 2, 1029 (1997).
- Crampton, M. R.; Rabbitt, L. C. Chem. Soc., Perkin Trans. 2, 1669 (1999).
- Crampton, M. R.; Rabbitt, L. C. Chem. Soc., Perkin Trans. 2, 2159 (2000).
- Norris, W. P.; Spear, R. J.; Read, R. W. Aust J Chem, 36, 297 (1983).
- Lowe-Ma, C. K.; Nissan, R. A.; Wilson, W. J.Org. Chem., 55, 3755 (1990).
- Fery-Forgues, S.; Vidal, C.; Lavabor, D. Chem. Soc., Perkin Trans. 2, 73 (1996).
- Kurbatov, S. V.; Budarina, Z. N.; Vaslyaeva, G. S.; Borisenko, N. I.; Knyazev, A. P.; Minkin, V. I.; Zhdanov, Yu. A.; Olekhnovich, L. P. Izv. Akad. Nauk. Ser. Khim., 1509 (1997).
- Evgenyev, M. I.; Garmonov, S. Y.; Evgenyeva, M. I.; Gazizullina, L. S. J. Anal. Chem., 53, 571 (1998).
- Terrier, F., Goumont, R.. ;. Pouet., M. J. ; Boubaker, T. ;. Halle., J.C., Polish Journal of Chemistry, 68, 2415 (1994).
- Terrier, F. ; Croisat, D. , Chatrousse, A.P. , Pouet., M. J. , Halle., J.C., Jacob, G., Journal of Organic. Chemistry, 57, 3684 (1992).
- Boughdiri, M. A. ; Fliss, O. ; Boubaker T. ; Tangour B., Oriental Journal of Chemistry, 22(3), (2006).
- Gaussian, Inc. Carnegie Office Park, Building 6 Pittsburgh, PA 15106 USA
- Barthelat, J. C, Ph. Durand, Chim. Ital, 108, 225 (1978).
- Barthelat, J. C., Molecular Physics, 65(2), 295 (1988).
- Carpenter, J. E.; Weinhold, F. , Mol. Struct. (Theochem) 169, 41 (1988).








![]()
![]()
