
The Role of Wnt Pathways in Neuropathic and Underexplored Pain Conditions: From Mechanisms to Treatment
 , Narayanan Jaisankar*and Chitra Vellapandian
, Narayanan Jaisankar*and Chitra Vellapandian Department of Pharmacology, Faculty of Medicine and Health Sciences, SRM Institute of Science and Technology, SRM College of Pharmacy, Kanchipuram, India
Corresponding Author E-mail: narayanj@srmist.edu.in
DOI : http://dx.doi.org/10.13005/bbra/3514
Download this article as:
![]()
Neuropathic pain is a chronic disease that occurs as a result of somatosensory damage, and is characterized by neuroinflammation, neuronal hyper excitability, and maladaptive plasticity. There is growing evidence that the Wnt signaling pathway is a key controller of these processes. Wnt3a mediates synaptic reorganization, glial activation, and release of pro-inflammatory cytokines via canonical Wnt/β-catenin signaling, and improves calcium influx, kinase activation, and central sensitization via non-canonical pathways, especially by Wnt5a and Ror2/Ryk receptors. In addition to neuropathic pain, abnormal Wnt signaling is also suggested to play a role in unexplored states of pain, such as visceral pain due to inflammation induced by obesity, cancer-related pain due to tumorigenic signaling, migraine vulnerability due to increased inhibitors like DKK1 and post-surgery pain because of fibrosis and compensatory repair. Anti-WNT3a/β-catenin (IWP-2, LGK974, XAV939), non-canonical, and anti-Ryk antibodies have demonstrated meaningful analgesic and neuroprotective effects in preclinical models. Taken together, these findings identify Wnt signaling as a dynamic mediator of pain sensitization and a compelling candidate for disease-modifying therapeutic strategies in neuropathic and chronic pain conditions.
KEYWORDS:Canonical Wnt pathways; Neuropathic pain; Non-canonical Wnt pathway; Pain; Wnt signaling pathways β-catenin
Introduction
Pain is an unpleasant affective and sensory experience that is linked or similar to tissue harm or possible tissue damage. Very often, pain is used as a symptomatic sign of a pathological condition or trauma. The successful treatment of the underlying pathology is central in such situations and it could result in the relief of the pain. However, the pain can persist even after the causative illness has been treated successfully and it can be because it is not possible to treat effectively the underlying medical condition, basically pain are two types acute pain and chronic pan.1 Acute pain has been described as the physiological reaction and subjective perception to noxious stimuli which can be pathologic, it normally shows a sharp onset and time constrained and acts to trigger avoidance behavior to reduce actual or potential tissue damage. Conversely, chronic pain refers to pain experienced during most days or daily, over the last three months or which causes functional limitation in at least one activity. Additionally, between 22 and 50% of general practitioner visits are due to chronic pain, making it a prominent reason for seeking medical attention.2,3 Depending on its cause, the pain might be categorized as nociplastic, neuropathic, or nociceptive. Multiple categories are often included in a single pain occurrence.Neuropathic pain may lead to increased pain sensitivity, including sudden pain, as well as sensory loss. Complex pathophysiological processes, such as altered endogenous pain modulation systems and increased excitability and ectopic activity in somatosensory nerve fibers, are involved in the underlying causes.4
Wnt signaling pathways are a collective of signal transduction cascades triggered by proteins which bind cell-surface receptors to transmit intracellular signals. Wnt signaling pathways rely on either same-cell (autocrine) or nearby cell-cell (paracrine) communication.5 The Wnt signaling pathway is a highly conserved and essential regulator of numerous cellular processes governing tissue homeostasis, cellular migration, proliferation, differentiation and embryonic development among other biological processes.6 Likewise, there are fewer findings that the development of neuropathic pain is influenced by Wnt signaling, which may also be a key mechanism behind the pathophysiology of neuropathic pain. Most types of a neuropathic pain grading system consist of four items: a pertinent clinical history, pain topography, sensory symptoms, and a diagnostic test that proves the presence of a neurologic lesion or illness. Depending on how many criteria are fulfilled, neuropathic pain may be classified as probable, definite or possible. This algorithm therefore classifies pain as possibly neuropathic if the neuroanatomical topography of the pain is plausible and The clinical history of the patient is in line with the occurrence of a neurological lesion or disease. In case the physical examination shows a sensory loss and/or hyperalgesia or allodynia in the respective area of the neuroanatomy, this also supports the diagnosis, in addition to the previously mentioned criteria, it is considered likely neuropathic. Lastly, neuropathic pain is only considered “definite” if an objective demonstration of a somatosensory system disease or lesion can be made.7 It is apparent that every Wnt ligand interacts with two different signaling pathways, namely, a non-canonical, β-catenin independent pathway, and the canonical, Wnt/ β-catenin pathway. However, a single Wnt ligand can activate multiple pathways, contingent on downstream component expression.8
The current review focuses on the role played by the Wnt signaling cascade in the pathogenesis of neuropathic pain and also looks at the role of Wnt signaling in pain related conditions. It highlights key molecular mechanisms by which Wnt components contribute to pain sensitization. The review also explores the involvement of Wnt signaling in pain-associated conditions. Furthermore, it discusses emerging therapeutic opportunities targeting this pathway. Overall, it aims to provide insights into Wnt driven pain mechanisms and potential intervention strategies.
WNT Signalling Overview
The Wnt gene family was sequenced and identified in the late 1900s by mice and Drosophila. In the case of mammary tumors, the MMTV often integrates into the same region of the mouse genome. They called this locus “int1” (short for “integration site 1”). Initially the int1 was not much known to be functional beyond mouse cancer, then in 1987 it was proved that the fruit fly (Drosophila melanogaster) has a highly conserved homolog of int1 in mice called “wingless (wg). This discovery was revolutionary since it revealed that int1 was a highly conserved developmental gene associated with cancer. This led to the naming of the broader Wnt gene (Wnt = Wingless + int1).9 The human Wnt gene, which is a single peptide resides on chromosome 12q13, codes encoding proteins with 23–24 fixed half sarcosine residues and an average length of 350–400 amino acids.10 There are approximately 100 known Wnt genes, which are divided into 13 subfamilies: It has been estimated that there are up to a hundred Wnt genes, which form thirteen subfamilies (Wnt1-Wnt11, Wnt16 and Wnt A). These genes can be functionally divided into two groups: the canonical Wnt signaling group (Wnt1, Wnt3a, Wnt3, Wnt7a, Wnt7b, and Wnt8) and the non-canonical one (Wnt4, Wnt5a, Wnt6, and Wnt11).11
There are other Wnt receptors beyond frizzled and LRP.Derailed/RYK transmembrane tyrosine kinase have a Wnt-binding domain that resembles those of the secreted Wnt-binding proteins.12 Derailed has been reported to interact with DWnt5 in Drosophila studies and hence regulate outgrowth of axons and migration of salivary glands.13,14 In C. elegans and mammals (RYK), these receptors also interact with Wnts. While RYK can interact with the Dishevelled (Dsh) protein to participate in the Wnt/beta-catenin/TCF pathway.15,16 ROR family members also have Wnt ligand binding capabilities via a cysteine-rich domain (CRD) similar to that of Frizzled receptors.17,18 This interaction is evolutionarily conserved, seen in organisms like C. elegans. For example, Wnt5a specifically binds to Ror2, inhibiting the Wnt/beta-catenin/TCF pathway, though the exact mechanism remains unclear.19 Xenopus experimental evidence shows that Wnt5a signaling through Ror2 controls cadherin expression and coordinates convergent extension motions. The receptor context paradigm stipulates that the final signaling is determined by the composition of receptors and not the ligand itself. E.g. Wnt5a whether complexed with Frizzled4 and LRP5 or complexed with ROR2 activates distinct downstream events, which are mediated by the Wnt/ β-catenin/TCF axis.20,21 Wnt signaling outcomes are not dictated by the Wnt ligand alone but by the specific receptor context. Different Wnts can bind to various receptors beyond Frizzled and LRP, such as RYK, ROR, and FRL1/Crypto, leading to diverse and sometimes conflicting pathway activations. This receptor-dependent specificity determines whether a Wnt will activate pathways like beta-catenin signaling or others, as seen with Wnt5a and Wnt11. Furthermore, co-receptors like Arrow can differentiate between Frizzled-mediated pathways, demonstrating that the entire receptor complex is crucial for specifying the final cellular response.22
Canonical Wnt/β-catenin pathway
Fig.1 demonstrates that the expression of the target genes is regulated in the nucleus by the canonical Wnt/ β -catenin signalling cascade. The assembly of axin, adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK3β), casein kinase 1α (CK1α), protein phosphatase 2α (PP2A), and the beta-transducin repeatcontaining ubiquitin protein ligase (β -transducin repeat -transcript complex) phosphorylate β -catenin in the absence of the presence of Wnt ligands. This phosphorylation directs β-catenin to be ubiquitinated and degraded by proteasomes. Where as the opposite happens when the Wnt proteins exist, as they bind the N-terminal cysteine-rich domain of Frizzled (Fzd) receptors, which attracts cytosolic disheveled proteins (Dsh in Drosophila and Dvl in mammals). This binding disrupts the formation of the APC/GSK3β/Axin complex and initiates Wnt signaling.
The Fzd receptors are a part of the G protein-coupled receptor based and have an archetypal seven-membrane structure. Additional other receptors and interactions between Fzd receptors and Wnt proteins are frequently required to spread Wnt signals efficiently. Among the notable co-receptors are receptor RTK like orphan receptor 2 (ROR2), lipoprotein receptor-related protein (LRP)-5/6, tyrosine kinase (RTK), TMEM59, Reck, and GPR124.23 Up on binding of Wnt to LRP5/6 and Fzd, the dephosphorylated Axin binds to the cytoplasmic tail of LRP5/6 and results in the phosphorylation and subsequent dismantling of the destruction complex. Although phosphorylation of Dvl inhibits the GSK3β destruction complex, which inhibits rendering β -catenin unstable, the dephosphorylation of Dvl decreases the stability and concentration of Axin. The outcome is that β-catenin builds up in the cytoplasm before moving to the nucleus and joining transcriptional coactivators such as BCL9, BRG1, CBP/p300 and Pygo, as well as TCF/LEF1, to start the transcription of Wnt target genes.After translocation into the nucleus, β -catenin activates gene transcription, which enhances neuronal excitability, inflammatory cytokines, and synaptic plasticity all are involved in the emergence and persistence of the nociceptive and neuropathic pain.24
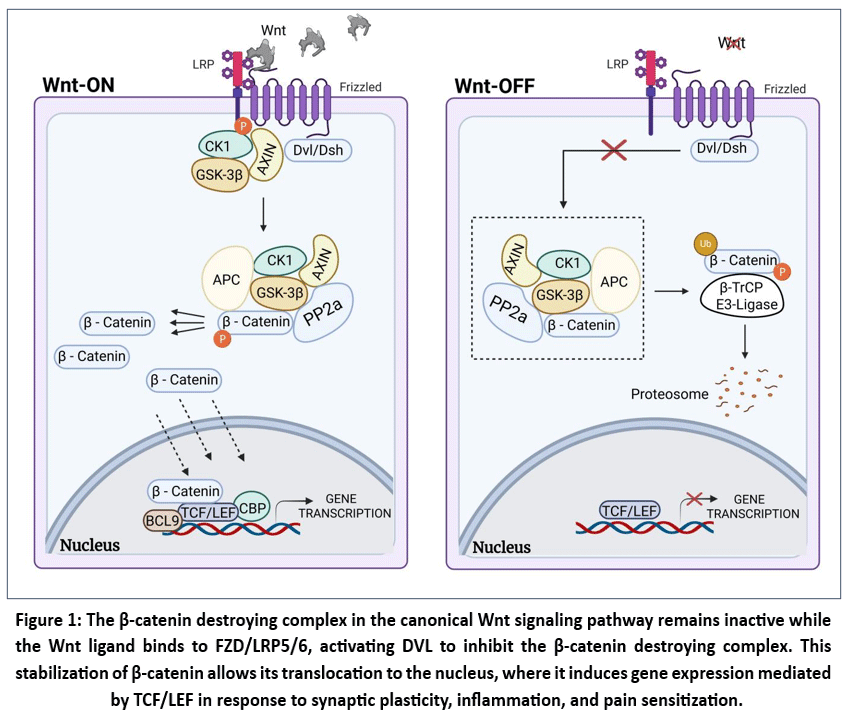 |
Figure 1: The β-catenin destroying complex in the canonical Wnt signaling pathway remains inactive while the Wnt ligand binds to FZD/LRP5/6, activating DVL to inhibit the β-catenin destroying complex.
|
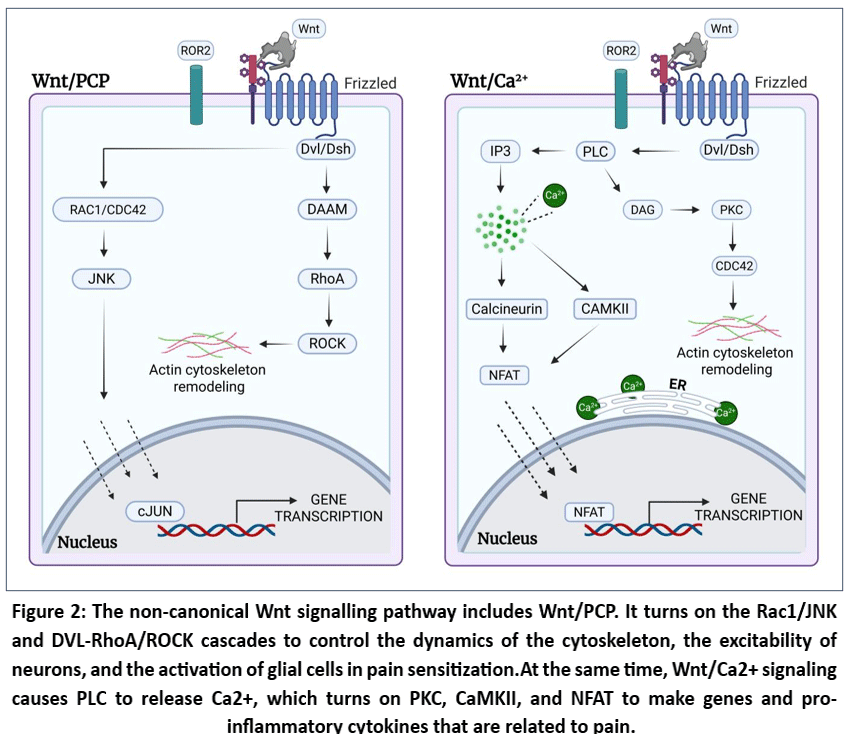 |
Figure 2: The non-canonical Wnt signalling pathway includes Wnt/PCP. It turns on the Rac1/JNK and DVL-RhoA/ROCK cascades to control the dynamics of the cytoskeleton, the excitability of neurons, and the activation of glial cells in pain sensitization.
|
NON-Canonical Wnt/β-catenin pathway
Fig 2, the non-canonical Wnt signaling pathway involves two large intracellular signaling cascades Wnt/calcium (Ca 2 + ) signaling pathway and Wnt/planar cell polarity (PCP) signaling pathway.25
2Wnt/planar cell polarity (PCP) pathway
Non-canonical Wnt signaling pathways, in contrast to the classic Wnt/beta-catenin signaling cascade, are independent of beta-catenin and crucial for regulating calcium homeostasis, polarity establishment, and other cellular processes. Wnt5a, Wnt7, and Wnt11 activate the planar cell polarity (PCP) pathway by binding to the Frizzled (Fzd) receptors and initiating downstream intracellular signaling by Dishevelled (Dvl/Dsh). This stimulation then leads to the stimulation of small GTPases like Rho and Rac that also stimulates Jun N-terminal kinase (JNK) through mitogen activated protein kinase (MAPK) signaling pathways.. There is, also, binding of Dvl/Dsh to Rho-associated kinase (ROCK) via DAAM1 (Dishevelled-associated activator of morphogenesis), thus amplifying downstream events.. Finally, it promotes cytoskeletal remodeling and increases the expression of ion channels by increasing the neuronal excitability (peripheral sensitization) correspondingly, it also increases the cytokines in the spinal cord and activates glial cells (central sensitization). Altogether, it contributes to aberrant and chronic propagation of neuropathic pain signals, and more and more experimental findings indicate that it plays vital role in the pathogenesis of neuropathic pain and its chronicity.26,27
Wnt/calcium (Ca2+) pathway
In the Wnt/calcium (Ca2+) pathway,Frizzled (Fzd) receptors are also bound to the Wnt ligands, such as Wnt1, Wnt5a, and Wnt11. and lead to activation of Dvl/Dsh and consequent activation of phospholipase C (PLC) via G protein coupled processes. The result of this activation is a rise in intracellular calcium, which contributes to the release of neuropeptides, activation of glial cells, and persistent neuroinflammation—all of which are strongly implicated in the development and maintenance of neuropathic pain.28-30 Besides, Dvl/Dsh has the ability to bind cGMP-specific phosphodiesterase 6, which results in decreased intracellular concentrations of cGMP and strengthening cytoplasmic calcium levels. The resultant calcium spike indirectly suppresses the canonical pathway by stimulating phosphorylation of TCF/LEF. Wnt/Ca²⁺is of especial significance to early embryonic development, neuronal communication, and inflammatory signaling.31
WNT in Neuropathic Pain
Role of wnt3a in Neuropathic Pain
An accumulating knowledge base points to the Wnt signaling pathway as a mediator of neuroinflammatory disease, glial activation, and synaptic plasticity involved in this state of pain. The impact of both canonical and non-canonical Wnt signaling and canonical on neuropathic pain could provide new targets to treat chronic pain.
Wnt3a is a canonical Wnt ligand that is central to neuropathic pain through activating β-catenin dependent transcriptional programs in the spinal cord. WNT3a expression rises in the dorsal horn following peripheral nerve damage and the interaction of Wnt3a with LRP5/6 co-receptors and Frizzled leads to inactivation of the β-catenin destroying complex permitting β-catenin buildup and nuclear translocation. This stimulation enhances the actions of genes associated with the potentiation of synapses, neuronal excitability, and glial activation, hence maintaining central sensitization. Also, Wnt3a increases excitatory neurotransmission by facilitating NMDA receptor activity and activating neurotrophic factors including BDNF, which amplifies maladaptive pain signaling. Hypersensitivity is inhibited by experimental blockage of Wnt3a/β-catenin signaling, which indicates its involvement as a mediator of neuropathic pain through a canonical pathway, and its importance as a therapeutic target.32,33
Mechanisms Linking Wnt Signaling to Pain Processing
Neuroinflammation
Abnormal Wnt/ β -catenin signaling activity leads to the development of a pro-inflammatory environment in the spinal cord.. Via β-catenin mediated translational programs in neurons and glial cells, Wnt signaling increases inflammatory cytokines such as IL-18, IL-1β and TNF-α. By encouraging astrocytic and microglial activation, this cytokine release improves central sensitization and nociceptive transmission.32
Microglia–BDNF Axis
The dorsal horn’s microglial cells are essential for NP. In vitro, Wnt3a promotes microglia to release BDNF after PSL damage, this mechanism is replicated in vivo. By modifying chloride ion gradients and down regulating the potassium-chloride co-transporter KCC2, BDNF is known to decrease inhibitory neurotransmission and increase pain transmission.33
Synaptic Plasticity and Central Sensitization
The amplification of pain signals depends on synaptic remodeling and plasticity, both of which are mediated by Wnt signaling. Wnt3a activation improves nociceptive transmission in CCI models by increasing the quantity and activity of excitatory synapses in DRG neurons and the spinal dorsal horn. CaMKII activation, elevated glutamate excitability, and Wnt-regulated trafficking of NMDA receptor subunits (such as NR2B) facilitate these effects.34
Reduction of Intraepidermal Nerve Fiber Density (IENFD)
Additionally, intraepidermal nerve fiber degeneration in peripheral tissues is facilitated by Wnt signaling. Wnt-mediated inflammatory and degenerative cascades partially regulate the loss of IENFD, which is seen in diabetic neuropathy and other peripheral neuropathies. This suggests that IENFD has a dual function in both central and peripheral sensitization.35
Role of wnt5a in neuropathic pain
The prototypical non-canonical Wnt ligand known as Wnt5a has been repeatedly implicated in pathology of neuropathic pain. It has been demonstrated that it is highly expressed in the area of superficial dorsal horn very after the nerve injury and is linked to neuronal excitability and synaptic remodeling, resulting in central sensitization.33 Such clinical relevance has been emphasized in the context of neuropathic pain induced by HIV gp120, where Wnt5a up regulation in the spinal dorsal horn is directly associated with mechanical allodynia, and where Wnt5a inhibition by, e.g., GAD67 gene therapy or pharmacological inhibition can notably reduce hypersensitivity.32
Mechanism of Action Linking Wnt5a to Pain Processing
Wnt5a acts, mechanistically, by activating the receptor tyrosine kinase Ror2 in the spinal dorsal horn and dorsal root ganglion. The activation of this receptor promotes non-canonical intracellular signaling cascades, the majority of which include phosphorylation of c-Jun N-terminal kinase (JNK) and Ca2+/calmodulin-dependent protein kinase II (CaMKII), which plays a major roles in expression of inflammatory genes and neuronal plasticity.32 These activities of kinases facilitate the release of pro-inflammatory cytokines such as tumor necrosis factor-alpha, interleukin-6 and interleukin-1B, which accelerates neuroimmune responses in the spinal cord., and increases pain hypersensitivity. Parallel Wnt5a signaling facilitates microglial activation and excitatory synaptic reorganization, which strengthens hyper excitability in spinal nociceptive pathways.33 Notably, these molecular and cellular changes are reversed by pharmacological inhibition of Wnt5a with Box5 or inhibition of its downstream kinases, which offers experimental support to Wnt5a-targeted interventions in neuropathic pain.32,34
Key Ligands and Receptors in Neuropathic Pain
Ligands: The most researched ligand linked to neuropathic pain is Wnt3a. Wnt5a is a non canonical ligand that has been linked to astrocytic activation and structural remodeling, while Wnt1 and Wnt7a are other canonical ligands.36
Receptors and Co-receptors: Co-receptors LRP5/6 and family of the Frizzled (Fzd) are the main receptors. In the canonical pathway, downstream proteins like β-catenin, Axin, GSK-3β, and Disheveled (Dvl) are essential transducers.37
Although β-catenin is known to be upregulated in the dorsal root ganglia and spinal dorsal horn in chronic pain models, its precise subcellular localization remains unclear. Alongside this, Wnt5a and its receptors Ror2 and Ryk are also markedly elevated following nerve injury in both peripheral and central compartments.38 Functionally, Wnt5a interacts with these atypical receptors to drive dendritic spine remodeling and synaptic excitability, while activating downstream effectors including CaMKII, NR2B, Src, and CREB through PCP and Wnt/Ca²⁺ signaling.38,39 All these signaling cascades facilitate neuroinflammation by means of IL-1 and TNF-alpha cytokines, enhance glutamatergic transmission, and aid in central sensitization.40 There is strong evidence showing the activation of the Wnt3α/ β-catenin signaling pathway in several rodent models of chronic pain including neuropathic pain.41-46
WNT in Less Studied Pain Conditions
Visceral Pain
Recent research suggests that non-canonical Wnt signaling, namely the Wnt5a/planar cell polarity (PCP) pathway, is essential for the pathophysiology of visceral discomfort by inducing inflammation in visceral adipose tissue (VAT). WNT5A expression is much higher in VAT than in subcutaneous adipose tissue (SAT) in obese people, and this is closely correlated with higher expression of intracellular mediators such as VANGL2 and PRICKLE1 as well as PCP receptors like ROR2 and FZD7. The downstream c-Jun N-terminal kinase (JNK) is an essential regulator of the inflammatory processes and malfunction in fat tissue, is activated by this Wnt5a-induced signaling cascade. Interestingly, WNT5A expression in VAT is associated with increased JNK activity and IL-6 production, a pro inflammatory cytokine that is strongly related to systemic low-grade inflammation and nociceptor sensitization, both of which are known to cause visceral hypersensitivity. In human VAT-derived preadipocytes, recombinant WNT5A markedly raised IL6 transcript levels in vitro, whereas WNT5A knockdown resulted in a 43% decrease in IL6 expression, indicating a direct regulatory connection. Crucially, these effects were considerably greater in diabetic participants, underscoring the way metabolic failure exacerbates Wnt-driven inflammatory pain pathways. Therefore, a novel molecular route connecting inflammation originating from adipose tissue to visceral nociceptive sensitization is represented by the WNT5A/PCP/JNK/IL-6 axis. This implies that Wnt5a targeted therapies may be clinically useful in the treatment of visceral pain disorders linked to obesity, such as diabetic gastropathy or irritable bowel syndrome (IBS), for the step by step overview of this sequence is mapped out in Fig. 3 below.47
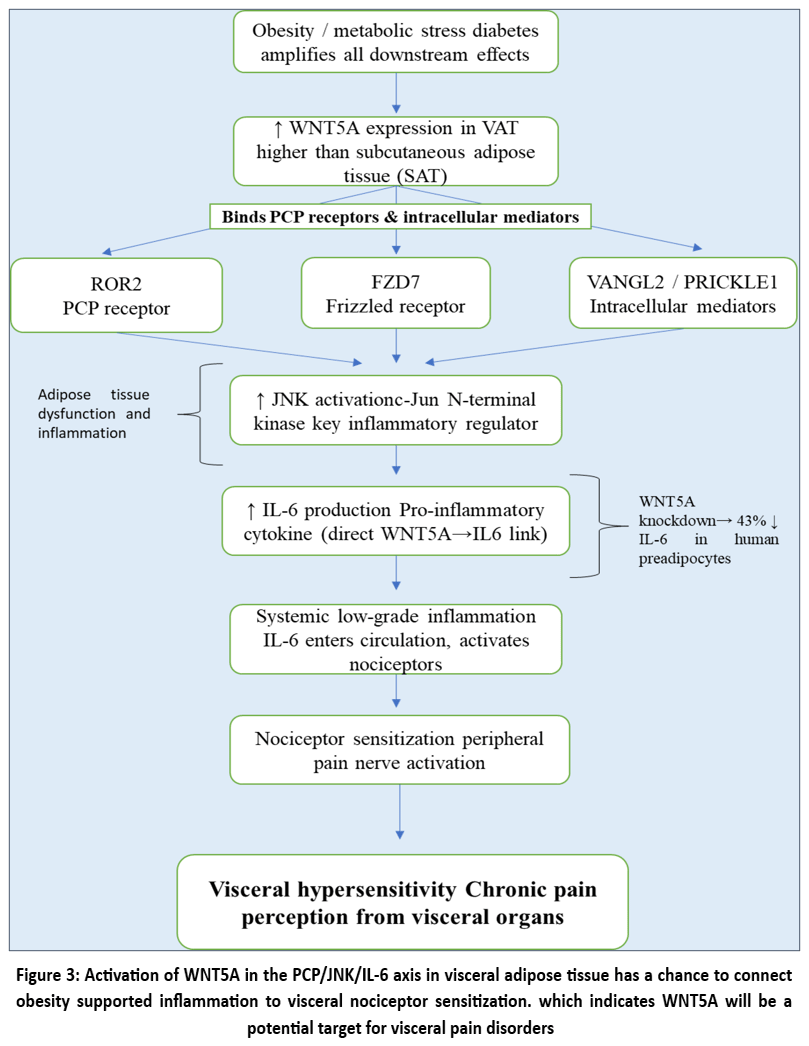 |
Figure 3: Activation of WNT5A in the PCP/JNK/IL-6 axis in visceral adipose tissue has a chance to connect obesity supported inflammation to visceral nociceptor sensitization.
|
Cancer and its link to chronic inflammation.
For the ease of understanding how cancer links to chronic inflammation is illustrated below Fig. 4.,wnt/beta-catenin signaling pathway one of the masterminds behind many human cancers, the Wnt/beta-catenin pathway is an important inducer and driver of these malignancies and a key risk factor that renders them resistant to treatment. In the canonical pathway, it involves the Wnt ligands (Wnt1, Wnt2, and Wnt3a), which bind to LRP5/6 receptors and Frizzled . This binding preserves β -catenin, and hence allows its translocation to the nucleus where it interacts with the TCF/LEF transcription factors.. This communication stimulates the expression of oncogenes such as Cyclin D1 (CCND1), c-MYC and MMP7, which facilitates cell proliferation, invasion, and metastasis. An important feature of this pathway is that it is often dysregulated in cancer. Gain-of-function mutations in the gene encoding β -catenin (CTNNB1) can constitutively activate Wnt signaling, and loss of function mutations of tumor suppressor genes, including APC and Axin, can do so. This is common in breast cancer, hepatic, and colorectal. In addition to tumor-promoting functions, Wnt pathway dysregulations have been linked to cancer stemness, chemo resistance, and oncogenicity in treatment-resistant hematologic, lung, and ovarian cancer. Moreover, non-canonical Wnt signaling, including the Wnt/Ca²⁺arms and Wnt/PCP, also plays roles in tumorigenicity through epithelial mesenchymal transition (EMT) and angiogenesis induction.48 The example of chronic pain and inflammation as observed in chronic pancreatitis or inflammatory bowel disease are well-documented risk factors of pancreatic and colorectal cancer, respectively.. In such conditions, inflammatory cues induce Wnt signaling activation in a pool of cells. This leads to a feedback mechanism in which Wnt-driven oncogenes further stimulate inflammation, resulting in malignant transformation. This link indicates that chronic pain, as an effect of persistent inflammation, may function as a catalyst for cancer due to the improper activation of a crucial signaling pathway.49
Migraine
The Recent genomic analyses have uncovered a compelling link between dysregulated Wnt/β-catenin signaling and migraine susceptibility, with key evidence pointing toward a causal role for elevated levels of Wnt pathway inhibitors, particularly Dickkopf-related protein 1 (DKK1). In this large-scale genome-wide association study (GWAS), 58 blood proteins was showing pleiotropic effects with migraine, of which DKK1 demonstrated a strong genetic causality (GCP = 0.88) for migraine risk. DKK1 is a well-known endogenous inhibitor of Wnt/β-catenin signaling, acting via LRP6 co-receptor antagonism to suppress β-catenin stabilization and transcriptional activity. Down regulation of Wnt signaling due to elevated DKK1 may disrupt neuronal homeostasis, brainstem integrity, and neurovascular coupling—mechanisms closely linked with migraine pathophysiology. Notably, the pons and medulla oblongata, areas linked to migraine aura and pain production, had matching RNA expression patterns for CGRP-related genes (CALCA) and DKK1. The study also found that platelet-derived growth factor B (PDGFB), which stimulates nociceptive nerve cells and can interact with Wnt-modulated pathways of vascular permeability and neuroinflammation, has a causal influence on migraine risk. When combined, our results suggest that reestablishing canonical Wnt signaling—possibly by boosting β-catenin activity or targeting DKK1 has the potential to support new treatment of migraines. Crucially, this work is the first to directly link disruption of the Wnt pathway to migraine in humans, bridging the gap among neurogenic, vascular, and metabolic pathways in chronic headache diseases and the details of the mechanisms have visually summarized below in Fig. 5.50
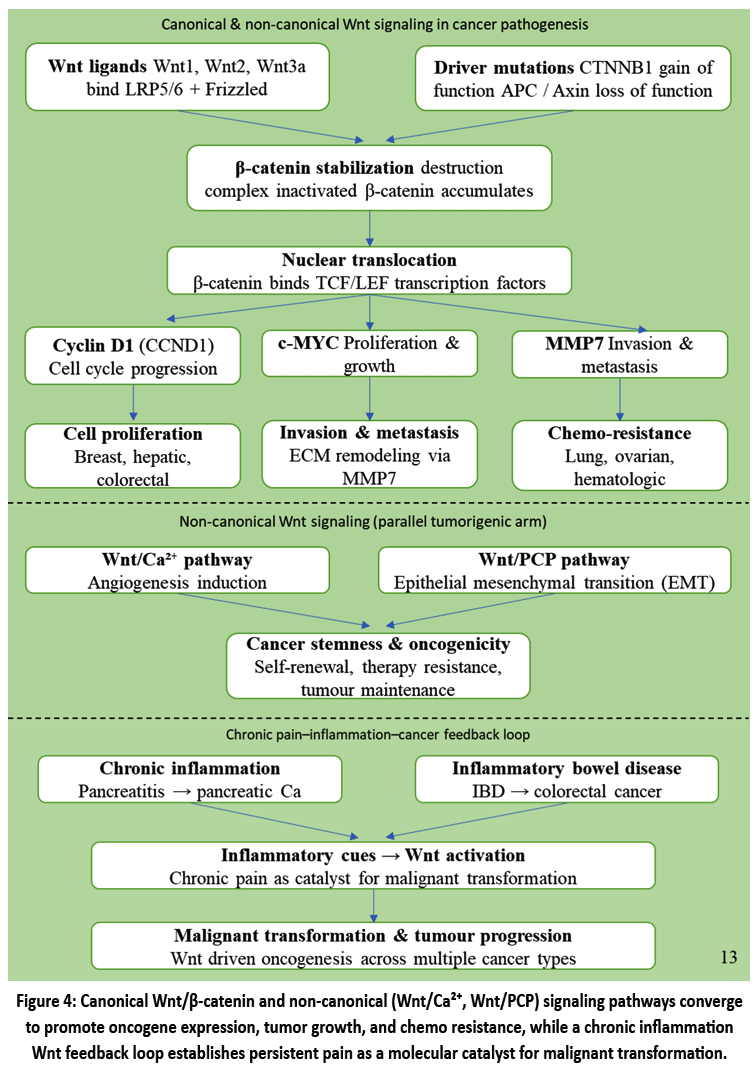 |
Figure 4: Canonical Wnt/β-catenin and non-canonical (Wnt/Ca²⁺, Wnt/PCP) signaling pathways converge to promote oncogene expression, tumor growth, and chemo resistance, while a chronic inflammation Wnt feedback loop establishes persistent pain as a molecular catalyst for malignant transformation.
|
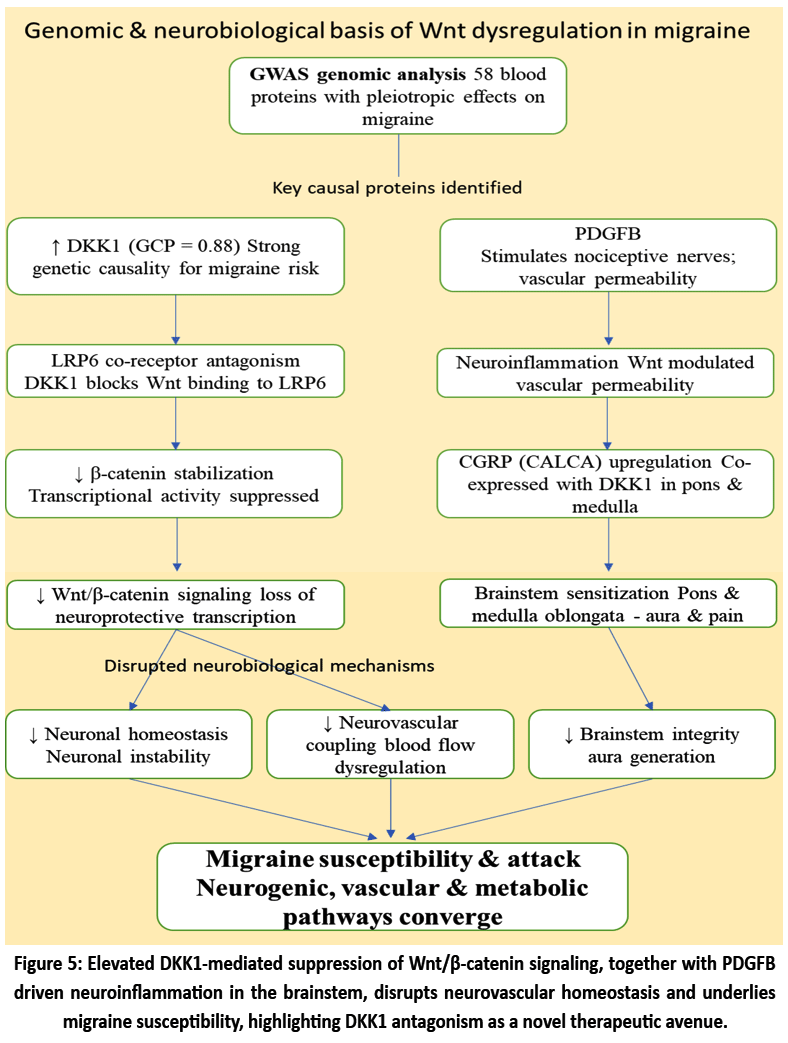 |
Figure 5: Elevated DKK1-mediated suppression of Wnt/β-catenin signaling, together with PDGFB driven neuroinflammation in the brainstem, disrupts neurovascular homeostasis and underlies migraine susceptibility, highlighting DKK1 antagonism as a novel therapeutic avenue.
|
Post Surgical Pain
The Fig. 6. outline the proposed condition of post surgical pain where according to new research, the Wnt signaling pathway may be involved in postoperative pain via processes including fibrosis, neuroinflammation, and neuronal sensitization. In research regarding corneal haze following photorefractive keratectomy (PRK), it is found that individuals who were more likely to have aberrant wound healing and fibrosis had considerable dysregulation of Wnt pathway components, including decreased expression of WNT3A and its downstream target SOX17. Increased inflammatory responses and extracellular matrix (ECM) remodeling, which are similarly linked to the pathophysiology of chronic pain, were linked to this downregulation.The connection between the Wnt pathway and TGF-β signaling, which is a significant modulator of neuronal hyperexcitability and fibrosis, suggests a potential connection with pain sensitization. For example, fibrotic markers like FN and ACAT2 were expressed more when TGF-β was activated in corneal epithelial cells. These markers may also play a role in nociceptive signaling at surgical sites. The study also demonstrated how PREX1, a RhoGEF that is increased in individuals at risk for fibrosis, promotes cell migration and extracellular matrix deposition, two mechanisms that might worsen mechanical discomfort after surgery and tissue stiffness. These results suggest that Wnt dysregulation may encourage a pro-fibrotic and inflammatory milieu, sustaining pain through stromal epithelial interactions and neural sensitization, even though there is currently no direct evidence connecting Wnt signaling to post-surgical pain. To determine if Wnt regulation might provide therapeutic advantages for post-surgical pain control, more investigation is required.51
Therapeutic Opportunities
Targeting Canonical Wnt Pathway with IWP-2 in Neuropathic Pain Management
In recent studies, targeting canonical Wnt signaling in neuropathic pain disorders has revealed good treatment potential. To test the action of IWP-2, a well-characterized Wnt-3a inhibitor, a chronic constriction injury (CCI) study to investigate it in a rat model of peripheral neuropathic pain, a well-known paradigm. Paw removal mechanical thresholds increased significantly after intrathecal treatment of IWP-2, suggesting a notable reduction in mechanical hypersensitivity. By lowering the expression of β-catenin, Frizzled-4, and Wnt-3a in the tissues of the dorsal root ganglia (DRG) and spinal cord, IWP-2 mechanistically inhibited the activation of the Wnt/β-catenin axis. This suppression stopped the downstream signaling cascade that involved the transcription of genes linked to synaptic plasticity, including those that control glutamate receptor activation, through β-catenin. This reduced the elevated activity of glutamatergic pathways, which are important causes of central sensitization. According to the analysis of Western blot, IWP-2 decreased the phosphorylation of CREB, CaMKII, PKC, NR2B, and Src—all of which are important mediators of increased pain sensitivity and synaptic transmission. In chronic neuropathic pain, these results establish canonical Wnt signaling as a promising therapeutic target and highlight the potential of IWP-2 as a disease-modifying analgesic.34 In conclusion, canonical Wnt/beta-catenin signaling inhibition by IWP-2 reduces neuronal hyperexcitability, suppresses pro-nociceptive signals, and notably restores mechanical allodynia in neuropathic pain models, raising the possibility that Wnt-3a/beta-catenin inhibitors can be developed as disease-modifying analgesics rather than symptomatic relief drugs.
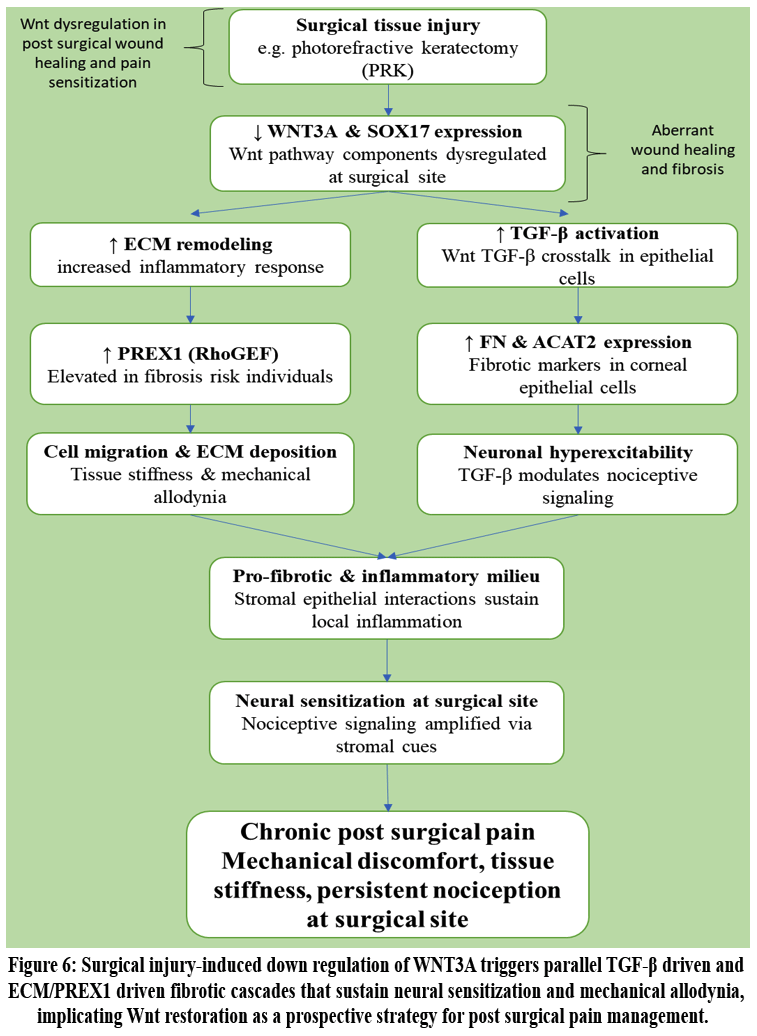 |
Figure 6: Surgical injury-induced down regulation of WNT3A triggers parallel TGF-β driven and ECM/PREX1 driven fibrotic cascades that sustain neural sensitization and mechanical allodynia, implicating Wnt restoration as a prospective strategy for post surgical pain management.
|
Therapeutic Inhibition of Non-Canonical Wnt/Ryk Signaling in Neuropathic Pain
Recent research has demonstrated that non-canonical Wnt/Ryk contributes to the pathogenesis of neuropathic pain, presenting a novel therapeutic targe. In a rodent model of chronic sciatic nerve injury, the spinal delivery of an anti-Ryk neutralizing antibody dramatically reduced neuropathic pain behaviors. By encouraging intracellular calcium influx and augmenting N-methyl-D-aspartate receptor sub type 2B (NR2B) activation, two essential components of synaptic plasticity and central sensitization, the Ryk receptor, A significant modulator of non-canonical Wnt signaling has been demonstrated to enhance neuronal hyper excitability. Blocking Ryk signaling lowered not only calcium-dependent signaling cascades but also the phosphorylation of synaptic proteins involved in excitatory transmission, eventually restoring mechanical and thermal pain thresholds..These results highlight how Wnt/Ryk-mediated pathways might be targeted therapeutically to reduce central sensitization and disrupt chronic pain networks in neuropathic pain disorders.50,51 In general, the blockade of Ryk, by inhibiting non-canonical Wnt/Ryk signaling, appears as a new therapeutic niche since targeting Ryk effectively decreases the influx of calcium, potentiation of the synapse, and nocifensive behavior characterized by pain hypersensitivity, providing a new perspective on how to block central sensitization in the molecular level of neuropathic pain.
Therapeutic Interventions Targeting Wnt Signaling in Chemotherapy-Induced Neuropathy
Inhibition of the Wnt/beta-catenin signaling pathway is a potential chemo-induced peripheral neuropathy (CIPN) treatment strategy. The results of preclinical studies support the effectiveness of blocking canonical Wnt signaling to prevent and reverse neuropathic pain due to chemotherapeutic drugs, such as paclitaxel. Intrathecal administration of multiple wnt signaling inhibitors, including LGK974 (a porcupine inhibitor), PNU74654 (β-catenin inhibitor), NSC668036 (a disheveled inhibitor), when administered intrathecally, greatly reduced mechanical and thermal hyperalgesia in CIPN rat models.These therapies did not only simply normalize thresholds but enhanced intraepidermal nerve fiber density (IENFD) and nerve conduction velocity, which suggested neuroprotective activity. Mechanically, this was linked to the down regulation of Dvl1, Wnt3a, c-myc, and \beta-catenin, as well as attenuated endoplasmic reticulum stress (GRP78) and neuroinflammation (MMP2) in the sciatic nerve tissue. Other works based on systemic dosing of LGK974, iCRT14 (inhibitor of β-catenin transactivation) and XAV-939 (β-catenin stabilization inhibitor) in rodents provide complementary results further supporting the therapeutic potential of the Wnt pathway inhibition. . These interventions also reduced the expression of Wnt3a, Wnt10a, in dorsal root ganglia (DRG), inhibited 8-catenin accumulation, and alleviated inflammatory markers including MCP-1, IL-1b. Histological studies showed that the β-catenin was localized in DRG neurons including CGRP-positive nociceptors and satellite glial cells, pointing to its key role in pain pathogenesis. Surprisingly, the preventive and post onset intervention with these inhibitors induced massive analgesic effects, and it is possible that blocking Wnt/beta-catenin signaling through pharmacological intervention may serve as a preventive and therapeutic intervention in CIPN with translational implications and ultimately impact the quality of life of patients undergoing neurotoxic chemotherapy.44,52 On balance, preclinical data strongly suggests that pharmacological inhibition of Wnt signaling not only relieves hyperalgesia in CIPN, but may also exert neuroprotective effects by preserving nerve architecture and decreasing inflammation, offering a highly translational approach to prevent chemotherapy-induced neuropathy in patients.
Discussion
The Wnt signaling pathway was a major regulator in our data of the induction and maintenance of neuropathic pain. The canonical (Wnt/beta-catenin), and the non-canonical (Wnt/Ca2+ and PCP) pathways are all up-regulated strongly in disease models of neuropathic pain to induce neuroinflammation, neuroplasticity, and synaptic plasticity, and glial cell excitability. In the canonical pathway, Wnt3a aggravates the pain signal by stimulating transcription of pro-inflammatory cytokines and pain-treated genes through Wnt3a- β-catenin signaling. At the same time, the non-canonical ligand, Wnt5a, binds to such receptors as Ryk and Ror2, triggering intracellular calcium influx, CaMKII and CREB activation, and dendritic spine rearrangement, which are also characterized by increasing pain transfer.
Disorders and diseases of neuropathic pain as diabetic neuropathy, post- surgical pain, chemotherapy induced neuropathy and visceral pain have demonstrated signs of putative failure in these pathways and aberrant activation of Wnt signalling components in both the peripheral and the central systems. Markedly, anti-inflammatory effects of Wnt inhibition have been observed in preclinical models of pain using Wnt ligand, Wnt receptor, Wnt downstream effector, IWP-2, LGK974, XAV939, or anti-Ryk antibodies, in which effects include reductions in pro-inflammatory mediators, intraepidermal nerve fiber reversal, and synaptic hyper excitability.
Alternative hypotheses and interpretive considerations
Pain is not always caused by too much Wnt– sometimes it is caused by too little. The migraine data involving DKK1 offers an important counterpoint to the idea that Wnt over activation is uniformly harmful. In migraine, elevated DKK1 suppresses Wnt/β-catenin signaling in the brainstem, and this loss of Wnt activity appears to destabilize neurovascular homeostasis and increase pain vulnerability. This means the relationship between Wnt and pain runs in both directions. A therapy designed to suppress Wnt in neuropathic pain could, in a different patient population, worsen migraine. This is a meaningful clinical concern, and it underscores the need to understand which direction Wnt is dysregulated before committing to a treatment strategy.
Shared molecular targets make it hard to credit Wnt alone. Several of the key downstream molecules implicated in Wnt-driven pain including CaMKII, CREB, NR2B, and JNK are also activated by entirely separate pathways such as TNF-α/NF-κB, TGF-β, and BDNF-TrkB signaling. In practical terms, this means that when a Wnt inhibitor reduces pain behavior in a rodent model, it is not always clear whether the effect is truly Wnt specific or whether the drug is simply dampening these shared effectors through an indirect route. This is an interpretive limitation that runs through much of the current preclinical literature and should be addressed through more targeted experimental designs, such as pathway-specific genetic knockouts alongside pharmacological interventions.
Wnt activation as a repair response that turns problematic over time. One reasonable interpretation of the early post injury data is that Wnt3a upregulation initially serves a repair purpose helping mobilize glial cells, support axonal regrowth, and restore disrupted synaptic connections. The problem may not be Wnt activation itself, but its failure to resolve. When this activation persists beyond the acute injury phase, it likely transitions from a healing signal into a driver of maladaptive sensitization. This matters clinically because broadly suppressing Wnt signaling, especially in conditions like diabetic neuropathy or chemotherapy-induced neuropathy where nerve regeneration is already impaired, could do more harm than good. A time-sensitive or phase-specific approach to Wnt modulation would be more appropriate than long-term pathway inhibition.
Limitations of the current evidence base
Reliance on animal models the Most mechanistic insights in this review come from rodent CCI and PSL models. While these are valuable and well-validated tools, they fall short of capturing the full complexity of human neuropathic pain, particularly its chronicity, psychological burden, and comorbidities. Rodent pain typically resolves within weeks, yet human neuropathic pain can persist for decades. Whether the Wnt dynamics observed in these acute models remain relevant in long standing human pain is a question the field has not yet answered.Translational delivery barriers are the most reviewed therapeutic studies relied on intrathecal drug delivery, effective experimentally but invasive and impractical for chronic outpatient use. Moving these findings toward real treatment options will require developing orally bioavailable, blood-brain barrier-crossing Wnt modulators or exploring targeted delivery platforms such as nanoparticle based systems. Poor selectivity of existing Wnt inhibitors. Agents like LGK974 and XAV939 suppress Wnt signaling system wide, raising safety concerns given Wnt’s essential roles in bone turnover, gut renewal, and immune regulation. Prolonged systemic inhibition risks bone loss, gastrointestinal toxicity, and impaired healing. Isoform-specific or spatially restricted inhibitors will need to be developed before Wnt targeted therapy becomes viable for chronic pain patients.Nevertheless, as mentioned in Section 6.2, major drawbacks such as dependence on animal testing and lack of specificity of existing Wnt blockers should be overcome in order to move these discoveries into a clinical setting
Conclusion
Neuropathic pain remains one of the most therapeutically challenging conditions in clinical medicine, and identifying its core molecular drivers is essential for developing effective, disease-modifying treatments. The review brings into attention the role of the Wnt signaling pathway that not only gets activated following injury but participates actively in the modulation of neuronal excitability, gliosis, synaptic plasticity, and neuroinflammation which contribute to chronic pain. Moreover, this signaling pathway is important for other less studied conditions. For example, there are studies indicating the importance of Wnt5a signaling in adipose tissue in nociceptor sensitization in visceral pain. Also, reduced canonical Wnt signaling via DKK1 expression may lead to disruption of neurovascular stability, thereby contributing to increased susceptibility to pain in migraines. In terms of cancer and post-surgical pain, there are different roles played by various Wnt pathways as they participate in inflammation and fibrosis leading to development of nociception. Regarding preclinical treatment, studies showed promising results from using inhibitors like IWP-2, LGK974, XAV939, as well as Ryk antibodies which not only helped with pain sensitivity but also protected nerves and decreased neuroinflammation.Taken together, the evidence reviewed here makes a strong case that the Wnt pathway is not merely a background regulator of neural development but an active, dynamic mediator of pain pathology, one that offers multiple, distinct points of therapeutic intervention across a broad spectrum of chronic pain conditions.Although further validation in humans will be required, based on the available preclinical data, Wnt signaling interference appears to be a promising approach in treating neuropathic and chronic pain.
Acknowledgement
The authors are thankful to the management of SRM Institute of Science and Technology, Kattankulathur campus for providing necessary facilities to carry out this work. The author further more acknowledges bio render.com for providing the tools and platform used in the creation of scientific illustrations for this manuscript.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable.
Author Contributions:
Chandaraa Kumar Pandiyan: Conceptualization, Investigation, Data curation, Formal analysis, Writing-original draft and visualization.
Narayanan Jaisankar: Conceptualization, Methodology, Data curation, Formal analysis, Writing-review and editing.
Chitra Vellapandian: Conceptualization, Supervision, Project administration
References
- International Association for the Study of Pain, 2025. Definitions of chronic pain syndromes. http://www.iasp-pain.org/advocacy/definitions-of-chronic-pain-syndromes/ (accessed 26 November 2025).
- Raja SN, Carr DB, Cohen M, et al. The revised International Association for the Study of Pain definition of pain: concepts, challenges, and compromises. Pain. 2020;161(9):1976-1982.
CrossRef - Spacek A. Modern concepts of acute and chronic pain management. Biomed Pharmacother. 2006;60(7):329-335.
CrossRef - Kang Y, Trewern L, Jackman J, McCartney D, Soni A. Chronic pain: definitions and diagnosis. BMJ. 2023;380:e076036.
CrossRef - Scholz J, Finnerup NB, Attal N, et al. The IASP classification of chronic pain for ICD-11: chronic neuropathic pain. Pain. 2019;160(1):53-59.
CrossRef - Nusse R, Varmus HE. Wnt genes. Cell. 1992;69(7):1073-1087.
CrossRef - Treede RD, Jensen TS, Campbell JN, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2008;70(18):1630-1635.
CrossRef - Finnerup NB, Haroutounian S, Kamerman P, et al. Neuropathic pain: an updated grading system for research and clinical practice. Pain. 2016;157(8):1599-1606.
CrossRef - Niehrs C. The complex world of Wnt receptor signalling. Nat Rev Mol Cell Biol. 2012;13(12):767-779.
CrossRef - Rijsewijk F, Schuermann M, Wagenaar E, Parren P, Weigel D, Nusse R. The Drosophila homology of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell. 1987;50(4):649-657.
CrossRef - Daniels DL, Eklof Spink K, Weis WI. β-catenin: molecular plasticity and drug design. Trends Biochem Sci. 2001;26(11):672-678.
CrossRef - Wong GT, Gavin BJ, McMahon AP. Differential transformation of mammary epithelial cells by Wnt genes. Mol Cell Biol. 1994;14(9):6278-6286.
CrossRef - Hsieh JC, Kodjabachian L, Rebbert ML, et al. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature. 1999;398(6726):431-436.
CrossRef - Yoshikawa S, McKinnon RD, Kokel M, Thomas JB. Wnt-mediated axon guidance via the Drosophila Derailed receptor. Nature. 2003;422(6932):583-588.
CrossRef - Harris KE, Beckendorf SK. Different Wnt signals act through the Frizzled and RYK receptors during Drosophila salivary gland migration. Development. 2007;134(11):2017-2025.
CrossRef - Inoue T, Oz HS, Wiland D, et al. C. elegans LIN-18 is a Ryk ortholog and functions in parallel to LIN-17/Frizzled in Wnt signaling. Cell. 2004;118(6):795-806.
CrossRef - Lu W, Yamamoto V, Ortega B, Baltimore D. Mammalian Ryk is a Wnt coreceptor required for stimulation of neurite outgrowth. Cell. 2004;119(1):97-108.
CrossRef - Kani S, Oishi I, Yamamoto H, et al. The receptor tyrosine kinase Ror2 associates with and is activated by casein kinase Iϵ. J Biol Chem. 2004;279(48):50102-50109.
CrossRef - Mikels AJ, Nusse R. Purified Wnt5a protein activates or inhibits β-catenin–TCF signaling depending on receptor context. PLoS Biol. 2006;4(4):e115.
CrossRef - Forrester WC, Kim C, Garriga G. The Caenorhabditis elegans Ror RTK CAM-1 inhibits EGL-20/Wnt signaling in cell migration. Genetics. 2004;168(4):1951-1962.
CrossRef - Schambony A, Wedlich D. Wnt-5A/Ror2 regulate expression of XPAPC through an alternative noncanonical signaling pathway. Dev Cell. 2007;12(5):779-792.
CrossRef - Tao Q, Yokota C, Puck H, et al. Maternal Wnt11 activates the canonical Wnt signaling pathway required for axis formation in Xenopus embryos. Cell. 2005;120(6):857-871.
CrossRef - Wehrli M, Dougan ST, Caldwell K, et al. Arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 2000;407(6803):527-530.
CrossRef - Martinez-Marin D, Stroman GC, Fulton CJ, Pruitt K. Frizzled receptors: gatekeepers of Wnt signaling in development and disease. Front Cell Dev Biol. 2025;13:1599355.
CrossRef - Xue C, Chu Q, Shi Q, Zeng Y, Lu J, Li L. Wnt signaling pathways in biology and disease: mechanisms and therapeutic advances. Signal Transduct Target Ther. 2025;10:106.
CrossRef - Holstein TW. The evolution of the Wnt pathway. Cold Spring Harb Perspect Biol. 2012;4(7):a007922.
CrossRef - Yang Y, Mlodzik M. Wnt-Frizzled/planar cell polarity signaling: cellular orientation by facing the wind (Wnt). Annu Rev Cell Dev Biol. 2015;31:623-646.
CrossRef - Girão H, Pereira P, Ramalho J, Quinlan R, Prescott A. Cholesterol oxides mediated changes in cytoskeletal organisation involve Rho GTPases. Exp Cell Res. 2003;291(2):502-513.
CrossRef - Chen Y, Chen Z, Tang Y, Xiao Q. The involvement of noncanonical Wnt signaling in cancers. Biomed Pharmacother. 2021;133:110946.
CrossRef - Flores-Hernández E, Velázquez DM, Castañeda-Patlán MC, et al. Canonical and non-canonical Wnt signaling are simultaneously activated by Wnts in colon cancer cells. Cell Signal. 2020;72:109636.
CrossRef - Ma L, Wang HY. Mitogen-activated protein kinase p38 regulates the Wnt/cyclic GMP/Ca²⁺ non-canonical pathway. J Biol Chem. 2007;282(39):28980-28990.
CrossRef - Kühl M, Sheldahl LC, Park M, Miller JR, Moon RT. The Wnt/Ca²⁺ pathway. Trends Genet. 2000;16(7):279-283.
CrossRef - Zhang YK, Huang ZJ, Liu S, Liu YP, Song AA, Song XJ. Wnt signaling underlies the pathogenesis of neuropathic pain in rodents. J Clin Invest. 2013;123(5):2268-2286.
CrossRef - Itokazu T, Hayano Y, Takahashi R, Yamashita T. Involvement of Wnt/β-catenin signaling in the development of neuropathic pain. Neurosci Res. 2014;79:34-40.
CrossRef - Zhao Y, Yang Z. Effect of Wnt signaling pathway on pathogenesis and intervention of neuropathic pain. Exp Ther Med. 2018;16:0.
CrossRef - Zhou YQ, Tian XB, Tian YK, Mei W, Liu DQ, Ye DW. Wnt signaling: a prospective therapeutic target for chronic pain. Pharmacol Ther. 2022;231:107984.
CrossRef - Tang Y, Chen Y, Liu R, Li W, Hua B, Bao Y. Wnt signaling pathways: a role in pain processing. Neuromolecular Med. 2022;24(2):233-249.
CrossRef - Fujita M, Demizu Y. Advances in the development of Wnt/β-catenin signaling inhibitors. RSC Med Chem. 2025;16(5):984-999.
CrossRef - Liu S, Liu YP, Huang ZJ, et al. Wnt/Ryk signaling contributes to neuropathic pain by regulating sensory neuron excitability and spinal synaptic plasticity in rats. Pain. 2015;156(12):2572-2584.
CrossRef - Simonetti M, Kuner R. Spinal Wnt5a plays a key role in spinal dendritic spine remodeling in neuropathic and inflammatory pain models and in the proalgesic effects of peripheral Wnt3a. J Neurosci. 2020;40(35):6664-6677.
CrossRef - Moyse E, Krantic S, Djellouli N, et al. Neuroinflammation: a possible link between chronic vascular disorders and neurodegenerative diseases. Front Aging Neurosci. 2022;14:827263.
CrossRef - Feng W, Teng R, Zhao Y, Gao J, Chu H. Epigenetic modulation of Wnt signaling contributes to neuropathic pain in rats. Mol Med Rep. 2015;12(3):4727-4733.
CrossRef - Lu K, Wang Q, Jiang H, et al. Upregulation of β-catenin signaling represents a single common pathway leading to the various phenotypes of spinal degeneration and pain. Bone Res. 2023;11:18.
CrossRef - Resham K, Sharma SS. Pharmacologic inhibition of porcupine, disheveled, and β-catenin in Wnt signaling pathway ameliorates diabetic peripheral neuropathy in rats. J Pain. 2019;20(11):1338-1352.
CrossRef - Resham K, Sharma SS. Pharmacological interventions targeting Wnt/β-catenin signaling pathway attenuate paclitaxel-induced peripheral neuropathy. Eur J Pharmacol. 2019;864:172714.
CrossRef - Shi Y, Yuan S, Li B, et al. Regulation of Wnt signaling by nociceptive input in animal models. Mol Pain. 2012;8:47.
CrossRef - Zhou X, Tao L, Zhao M, et al. Wnt/β-catenin signaling regulates BDNF release from spinal microglia to mediate HIV-1 gp120-induced neuropathic pain. J Neuroinflammation. 2020;17:107.
- Zuriaga MA, Fuster JJ, Farb MG, et al. Activation of non-canonical WNT signaling in human visceral adipose tissue contributes to local and systemic inflammation. Sci Rep. 2017;7:17326.
CrossRef - Najafi SMA. The canonical Wnt signaling (Wnt/β-catenin pathway): a potential target for cancer prevention and therapy. Iran Biomed J. 2020;24(5):264-275.
CrossRef - Tanha HM, Nyholt DR. Genetic analyses identify pleiotropy and causality for blood proteins and highlight Wnt/β-catenin signalling in migraine. Nat Commun. 2022;13:2593.
CrossRef - Yu F, Yu C, Li F, et al. Wnt/β-catenin signaling in cancers and targeted therapies. Signal Transduct Target Ther. 2021;6:307.
CrossRef - Kumar NR, Khamar P, Shetty R, et al. Identification of novel predictive factors for post-surgical corneal haze. Sci Rep. 2019;9:16980.
CrossRef
Abbreviations
APC: Adenomatous polyposis coli
BDNF: Brain-derived neurotrophic factor
CaMKII: Calcium/calmodulin-dependent protein kinase II
CCI: Chronic constriction injury
CK1α: Casein kinase 1 alpha
CREB: cAMP response element-binding protein
DKK1: Dickkopf-related protein 1
DRG: Dorsal root ganglion
Dvl/Dsh: Dishevelled protein
ECM: Extracellular matrix
EMT: Epithelial–mesenchymal transition
Fzd: Frizzled receptor
GSK-3β: Glycogen synthase kinase-3 beta
IBS: irritable bowel syndrome
IENFD: Intraepidermal nerve fibre density
IL-1β / IL-6 / IL-18: Interleukin-1 beta / 6 / 18
JNK: c-Jun N-terminal kinase
LRP5/6: Low-density lipoprotein receptor-related protein 5/6
MAPK: Mitogen-activated protein kinase
NMDA: N-methyl-D-aspartate
NP: Neuropathic pain
PCP: Planar cell polarity
PKC: Protein kinase C
PLC: Phospholipase C
PRK: Photorefractive keratectomy
PSL: Partial sciatic nerve ligation
ROR2: Receptor tyrosine kinase-like orphan receptor 2
RTK: Receptor tyrosine kinase
RYK: Related to tyrosine kinase
SAT: Subcutaneous adipose tissue
TCF/LEF: T-cell factor / lymphoid enhancer-binding factor
TGF-β: Transforming growth factor beta
TNF-α: Tumour necrosis factor alpha
VAT: Visceral adipose tissue
Wnt: Wingless-related integration site
Accepted on: 09-05-2026
Second Review by: Dr. Akshaya Arva
Final Approval by: Dr. Wagih Ghannam








![]()
![]()
