
Osteochondral Unit Dysfunction in Osteoarthritis: Genetic, Epigenetic, and Hormonal Mechanisms
 , Annapareddy Bindusha2and Boyapally Maheshwari Reddy3*
, Annapareddy Bindusha2and Boyapally Maheshwari Reddy3*
1Department of Pharmacy Practice, Faculty of Engineering and Technology, University Institute of Pharmaceutical Technology, Annamalai University, Tamilnadu, India.
2Department of Pharmacy Practice, Joginpally B.R. Pharmacy College, Hyderabad, India.
3Department of Pharmacology, Joginpally B.R. Pharmacy College, Hyderabad, India.
Corresponding Author E-mail: mahi.unaj@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3511
Download this article as:
![]()
Osteoarthritis (OA) is a common and debilitating musculoskeletal condition that is associated with a progressive degeneration of joints and persistent pain. OA has recently been identified as a whole-joint condition characterized by a coordinated pathological process affecting cartilage, subchondral bone, synovium, and periarticular tissues. There is growing evidence of the importance of subchondral bone remodelling, disrupted joint biomechanics, and molecular crosstalk between bone and cartilage in the pathogenesis of disease. This review summarises about the genetic, genomic, epigenetic, and hormonal variables that contribute to osteoarthritis and more specifically how they selectively control osteochondral homeostasis.A comprehensive literature search was conducted using electronic databases including PubMed, Scopus, and Web of Science. Articles published between 2000 and 2025 were screened. Studies focusing on genetic, epigenetic, and hormonal mechanisms involved in osteochondral dysfunction were selected based on relevance and predefined inclusion criteria.There are several variants of the interactions with cartilage matrix and bone remodelling, and inflammatory signaling, which are being identified by genetic studies, and epigenetic mechanisms, such as DNA methylation, histone modifications, and non-coding RNAs are viewed as having a central role in controlling the expression of genes in joint tissues. The skeletal metabolism is further controlled by hormonal factors, including estrogen, parathyroid hormone, growth hormone-IGF-1, and adipokines, which determine the OA susceptibility especially in old age and postmenopausal stages. Besides that, the development of the bone density measurement, such as high-resolution imaging, biochemical biomarkers, epigenetic profiling, artificial intelligence-based imaging, and multi-omics, is broadening the concept of bone quality beyond traditional measures.
KEYWORDS:DNA methylation; Epigenetic; Histone modifications; Osteoarthritis; Subchondral bone
Introduction
Osteoarthritis (OA) is the most widespread type of arthritis, as well as a significant source of chronic pain, loss of functionality, and disability in the world, especially in older adults. Even though OA was previously considered a disease that developed mainly due to the degeneration of the articular cartilage, nowadays, it is becoming increasingly evident that OA is a whole-joint disease that includes pathological alterations in articular cartilage, subchondral bone, synovium, ligaments and periarticular muscles.1,2 Changes in subchondral bone density, microarchitecture and mechanical properties are also among these tissues whose changes are becoming popularly known as important determinants of joint biomechanics and disease progression.
OA is no longer considered to be a condition that is caused by mechanical wear and tear only. Rather, it is known to be a multifactorial, heterogeneous and complex disease that is defined by interaction of genetic disposition with environmental factors.3The paradigm shift has led to massive studies on the pathogenesis of OA at a molecular level, with a focus on the genetic and genomics and epigenetics. Osteoarthritis, in its clinical and pathological manifestation, is a scenario of degenerative and inflammatory processes in the synovial joints in the form of osteophytes, hyperplasia and inflammation of the synovium, as well as progressive loss of articular cartilage that leads to pain, stiffness, and loss of joint function.4 Although this progress has been made in comprehending OA as a multi-tissue disease, temporal and causal connections of pathological alterations of individual joint tissues are yet to be established fully. There is still no evidence as to whether the primary changes that occur in subchondral bone trigger secondary changes in cartilage and synovium or whether they change simultaneously.5Along with traditional risk factors like old age, obesity, joint trauma, mechanical overload, there has been significant evidence to indicate that genetic, genomic, epigenetic, and hormonal factors play an important role in disease susceptibility and progression.6,7
Genetic research has confirmed that there are many DNA variants that are linked with the risk of OA, highlighting the role played by genes in the development of the disease.8Genetic factors can have direct impacts on the joint tissues, as in disease like Stickler syndrome where dysfunctional mutations in COL2A1 result in defective type II collagen synthesis and premature joint degeneration.9Alternatively, genetic variants can exert an indirect effect because they can disable the repair and maintenance of cells by aberrantly regulating gene transcription.10 Hypotheses in emerging research also explain how evolutionary mismatches in the ancestral genetic variation and the dynamic environments in the modern world may predispose individuals to OA as a mismatch disease. These findings have been enhanced by genomic studies, which have helped in explaining the molecular pathways and mechanisms of cartilage homeostasis, extracellular matrix remodelling, inflammation, and bone turnover.
The epigenetic regulation, including DNA methylation, chromatin modification, and non-coding RNAs (ncRNAs), is essential in regulating gene expression but does not affect a sequence of DNA. Epigenetic regulation plays a crucial role in maintaining normal functioning and homeostasis of cells and tissues, and its impairment is becoming more apparent in OA. Distorted forms of epigenetic patterns have been linked to changes in the expression of genes that mediate cartilage degeneration, inflammatory signalling, and subchondral bone remodelling. Besides enhancing the knowledge on OA pathogenesis, epigenetic modifications have the potential to be used as biomarkers to stratify, prognosticate, and give responses to diseases.
Hormonal determinants are important in the regulation of the risk and progression of osteoarthritis (OA) on top of genetic and epigenetic regulators. The increased occurrence of OA among postmenopausal women supports the relevance of estrogen in the maintenance of joints. Estrogen controls the metabolism of cartilage, subchondral bone remodelling, synovial inflammation, and its decreases encourages cartilage degeneration, distorted bone turnover and inflammation. Anabolic and catabolic processes of joint tissues are also affected by other hormonal systems, such as the GH -IGF axis, thyroid hormones and metabolic hormones, including leptin and insulin. The variability in OA susceptibility and severity of the disease is due to dysregulation of these pathways.
This review provides a critical synthesis of recent data on the implication of genetics, genomics, epigenetics, and hormonal factors in osteoarthritis and explains how these interdependent regulatory processes can be used to induce dysfunction in joint tissues and disease progression.
Literature Search Strategy
A narrative literature review was performed using electronic databases such as PubMed, Scopus, and Web of Science. Keywords including “osteoarthritis,” “osteochondral unit,” “genetics,” “epigenetics,” “DNA methylation,” “hormonal regulation,” and “subchondral bone” were used in various combinations. Peer-reviewed articles published primarily in the last two decades were considered. Landmark older studies were included where scientifically relevant. Studies not available in English or lacking sufficient methodological clarity were excluded.
Subchondral Bone in OA Pathophysiology
Reasonable comprehension and intervention for OA should focus on indivisible levels from the organism scope of resolution because it results from a coupled pathological action of joint tissues with systemic regulators, not just simple degeneration of cartilage. At tissue level, mechanical microdamage and Biomechanical loading in subchondral bone under wiredrawn induces adaptive remodelling with increased turnover, altered mineralization and sclerosis. These alterations change the biomechanical environment of the overlying cartilage, becoming stiffer with less shock absorption, leading to accelerated cartilage matrix degeneration.
Meanwhile, there is a biochemical crosstalk between subchondral bone and cartilage that serves as the hub of OA evolution. Activated osteoblasts and osteoclasts in diseased bone produce a variety of growth factors, cytokines, and signalling molecules including transforming growth factor-β (TGF-β), Wnt ligands, and bone morphogenetic proteins (BMPs).
These mediators diffuse through the osteochondral junction and disturb chondrocyte homeostasis by inducing hypertrophic differentiation, extracellular matrix degradation, and diminished anabolic repair.
The synovium also enhances the progress of disease with a peripheral low-level chronic inflammation. Pro-inflammatory cytokines, chemokines and enzymes responsible for matrix degradation are produced by synovial macrophages and fibroblasts in this process, which result in the enhancement of cartilage catabolism, as well as an impact on bone remodelling. This inflammatory environment sets up a vicious cycle of tissue damage inducing more inflammation and degradation spanning the entire joint.
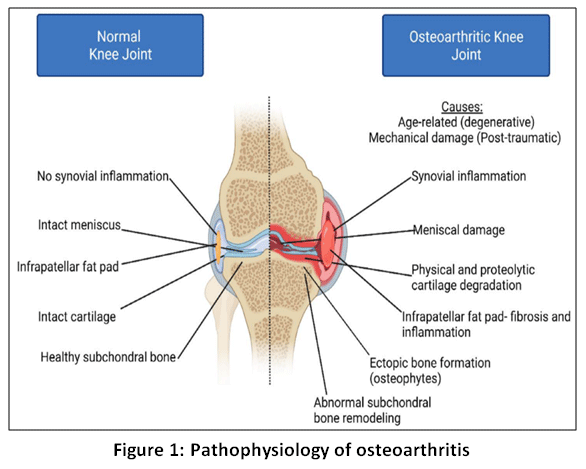 |
Figure 1: Pathophysiology of osteoarthritis
|
Osteoarthritis (OA) is a complex disease entity involving articular cartilage, synovium, infrapatellar fat pad and the subchondral bone. OA is characterized by chronic inflammation, degradation cartilage mechanical and proteolytic as well as aberrant subchondral bone formation that extend beyond the normal joint margins in the form of bony outgrowths into the joint capsule or periarticular soft tissues, called osteophytes.
Genetic Determinants of Osteoarthritis
Genetic determinants impact osteoarthritis (OA) pathogenesis by modulating fundamental biological pathways in joint development, maintenance, and repair. Heritability estimates demonstrate that approximately 40–65% of the risk for OA is determined by genetics, with a stronger genetic effect on hip and hand compared to knee OA.11,12 Genetic variants influence several joint tissues concomitantly, underlying OA as a whole joint disorder.
Variants in genes encoding extracellular matrix components and regulatory factors, like COL2A1 and GDF5, affect chondrocyte differentiation, collagen organisation, and proteoglycan synthesis at the cartilage level.13 These changes make cartilage more vulnerable to matrix degradation and less resilient to mechanical loading. Simultaneously, genetic variation in TGF-β pathway components and signalling molecules like SMAD3 disturbs chondrocyte homeostasis, encouraging hypertrophic differentiation and upregulating matrix-degrading enzymes like aggrecanases and matrix metalloproteinases.14
Subchondral bone remodelling is additionally influenced by genetics. Sclerosis and aberrant load transmission throughout the osteochondral unit result from variations that impact osteoblast and osteoclast activity, which also change bone turnover, mineralisation, and microarchitecture. Cartilage deterioration is further accelerated by this mechanically unfavourable environment. Furthermore, synovial responses are modulated by genes related to immune and inflammatory regulation, which affects cytokine production and low-grade inflammation that exacerbates joint tissue damage.
Furthermore, disease heterogeneity is shaped by interactions between genetic determinants, ageing, mechanical stress, and metabolic factors. OA-associated genetic variations predispose joints to progressive degeneration under physiological loading conditions by decreasing the threshold for tissue damage and impairing repair capacity, rather than functioning as isolated triggers.15
Although osteoarthritis (OA) affects many joint tissues, cartilage degradation continues to be the pathological hallmark, particularly in more advanced stages of the disease. Since collagen, proteoglycans, and water make up the majority of articular cartilage, genes that encode cartilage structural elements have been extensively investigated as potential risk factors for OA. The main collagenous framework of articular cartilage is type II collagen, which is encoded by the COL2A1 gene on chromosome 12. Rare Mendelian disorders with early-onset OA, such as Stickler syndrome, chondrodysplasias, and Kniest dysplasia, have been found to be associated with pathogenic mutations in COL2A1.16 Multiple linkage and association studies have established associations between COL2A1 variants or haplotypes and generalised or early-onset OA across different groups of people, regardless of the fact that COL2A1’s contribution to common OA is less consistent.17These results suggest that COL2A1 variants are especially important in early-onset, multi-joint OA, where genetic testing may be taken into consideration, though more research needs to be conducted to determine its predictive value and cost-effectiveness.
Aggrecan, which is encoded by the ACAN gene on chromosome 15, builds up a majority of proteoglycans in articular cartilage. ACAN mutations have been linked to idiopathic short stature, spondyloepiphyseal dysplasia, and familial osteochondritis dissecans—disorders commonly linked to early OA.18 Large-scale linkage analyses and GWAS have not revealed a consistent relationship between ACAN and common idiopathic OA across joint locations,19indicating a limited function for ACAN in typical late-onset OA, despite its obvious relevance in hereditary skeletal illnesses.
Asporin, a cartilage extracellular matrix protein, is encoded by the ASPN gene located on chromosome 9. It plays a role in collagen binding and mineralization, helping to maintain cartilage structure and function. Through functional studies, it has been shown that certain ASPN alleles inhibit receptor-mediated signaling from TGFβ, resulting in decreased expression of key cartilage matrix genes such as COL2A1 and ACAN, thus limiting the accumulation of proteoglycans during chondrogenesis. Genetic association studies have shown that the association between ASPN alleles and OA severity is different across ethnic groups, although the association is not consistent in Caucasians.20 This suggests that the role of ASPN in OA susceptibility may vary depending upon ethnicity.
Collagen IX and collagen XI, encoded by COL9A1 and COL11A1, respectively, play an important role in the structural organization and mechanical integrity of cartilage through the regulation of type II collagen fibril assembly and spacing. While mutations in these genes result in rare forms of skeletal dysplasia,21 association studies suggest that they may contribute to the development of OA in more complex ways. While COL9A1 has mainly been connected with OA of the hip, suggesting that specific genes will only affect certain joints; COL11A1 is the strongest, most consistent gene associated with cartilage structure involved in OA, as evidenced by GWAS, Meta-Analyses, and multiple Population Studies through differing ethnicities. Therefore, the information collected shows that rare, high-impact mutation in the cartilage matrix genes has a substantial effect on Early-onset OA as well as Familial OA; while Common OA results from the combined effect of many genes with modest and sometimes Contextually dependent effects on the biology of the OA based on an individual’s genetics and the biomechanical properties of their joints within a given population.
As the matrix begins to break down due to genetic changes in cartilage structure, it is now accepted that osteoarthritis affects not only the cartilage itself but also other parts of the osteochondral unit. The articular cartilage depends heavily on the structural properties of the subchondral bone for support and effective distribution of forces at the joint surface. Additionally, the subchondral bone also plays a role in mediating the biochemical interaction between chondrocytes and the structural support of articular cartilage. In conclusion, any genetic change at the level of the cartilage matrix that weakens its architecture will result in an altered joint structure and will ultimately result in osteoarthritis.
Molecular Pathways Linking Cartilage Genetics and Subchondral Bone Remodeling
Dysregulation of multiple conserved signaling pathways mediates both directions of ecological interaction between the joint and the surrounding area, including subchondral/epiphyseal regions and articular cartilage. This coordination is critical for maintaining normative homeostatic equilibrium and promoting OA. The Wnt/β-catenin signaling pathway is specifically related to the differentiation into chondrocytes and osteoblasts. Maintenance of collagenous tissue (articular cartilage) requires the presence of moderately activated Wnt signaling. Increased Wnt activation initiates the hypertrophy of chondrocytes and excessive degradation of the matrix, along with the overproduction of subchondral bone. All manipulate articular cartilage and lead to thinning of articular cartilage and sclerosis of subchondral bone; genetic polymorphisms that alter the regulators of the Wnt signaling pathway can have a simultaneous effect (positive or negative) on both the integrity of articular cartilage and the morphology and mass of subchondral bone.
The important role played by TGF-β within the articular cartilage/subchondral bone (osteochondral unit) can be divided into two parts: it maintains the phenotype of chondrocytes and does not allow terminal differentiation of these cells; on the other hand, when TGF-β is inappropriately activated due to excess mechanical overload or matrix defects, this TGF-β produces abnormal signalling leading to increased numbers of osteoblasts being recruited and increasing the activity of osteoblasts producing new bone Localized elevation of TGF-β activity in subchondral bone promotes sclerosis and osteophyte formation, further accelerating cartilage degeneration. Bone morphogenetic proteins (BMPs), members of the TGF-β superfamily, regulate synthesis of the extracellular matrix and skeletal development. Physiological signalling through BMPs promotes repair of cartilage and formation of new bone; however, if BMP activity is dysregulated, chondrocytes hypertrophy and form osteophytes. Genetic predisposition toward producing or responding to BMP ligands and antagonists can also play a role in shifting the balance of tissue growth and repair toward pathogenic (pathological) remodelling in both cartilage and subchondral bone.
The RANKL-OPG axis regulates the formation and resorption of bone by regulating the differentiation and activity of osteoclasts. Elevated levels of RANKL and declined levels of OPG lead to increased subchondral bone resorption during early stages of OA, followed by compensatory bone formation at later stages and subsequently sclerosis of the subchondral bone. Changes in signals derived from cartilage that are mediated by mutations in genes encoding for matrix components can also influence RANKL-OPG signaling, which connects the breakdown of cartilage with the changing architecture of subchondral bone.22
These pathways illustrate how genetic changes in cartilage structure can result in the initiation of a sequence of molecular changes that will alter the biology of subchondral bone.23 The relationship between various signaling molecules like Wnt, TGF-β, BMP, and RANKL-OPG indicates that the osteochondral unit is a coordinated system and that for effective disease-modifying therapies for OA, both cartilage deterioration and subchondral bone remodeling need to be targeted.
Epigenetic Factors Affecting Bone Density
DNA methylation
Covalently attaching a methyl group (CH₃) is an epigenetic mark known as DNA methylation whose purpose is to regulate transcriptional expression of genes. DNA methylation adds a methyl group (CH₃) to the carbon 5 site of cytosine, typically on CpG sites within regulatory regions (CpG islands) of gene promoters. Gene regulation via DNA methylation is a significant mechanism of transcriptional control and it also functions to regulate gene expression during the process of bone remodelling by affecting the differentiation of both osteoblasts (bone forming cells) and osteoclasts (bone resorbing cells). Increased levels of DNA methylation within promoter CpG islands of specific transcription factors (RUNX2, OSX (SP7), ALPL, COL1A1) in osteogenic cell populations leads to decreased transcriptional expression of these factors resulting in less osteoblast differentiation, reduced formation of extracellular matrix and decreased mineralization of bone. On the other hand, a reduction in the level of DNA methylation (hypomethylation) within the genes involved in osteoclast differentiation (RANKL, NFATc1) leads to increased differentiation of osteoclasts and enhanced bone resorption resulting in an imbalance of the skeleton.24,25The regulation of DNA methylation patterns comes from the activity of the DNA methyltranferases (DNMTs), which control and maintain the patterns that have formed.
DNMT1 is primarily responsible for maintenance methylation during the DNA replication cycle. It identifies hemi-methylated DNA and methylates the new strand to restore symmetrical methylation of DNA, allowing for the preservation of epigenetic information (gene methylation patterns) when cells divide. This is crucial for maintaining lineage-specific gene expression patterns in bone cells. DNMT3A and DNMT3B serve as de novo methyltransferases by adding methyl groups to unmethylated CpG sites during the early stages of development and differentiation of cells. These enzymes establish the appropriate tissue-specific DNA methylation landscape and provide precise regulation of gene expression to facilitate osteogenic commitment and skeletal development.
DNA methylation is also reversible in a dynamic fashion through the function of ten-eleven translocation (TET) dioxygenases (TET1, TET2, and TET3). The TET family enzymes initiate the process of active demethylation by oxidizing 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), which can then be converted to 5-formylcytosine (5fC) and finally to 5-carboxylcytosine (5caC), resulting in demethylation of 5mC and 5hmC after base excision repair. Gene methylation is dynamic, enabling precise control of gene expression along with developmental cues, aging, hormonal changes, oxidative stress, and inflammatory stimuli through combining the functions of the DNMT family and TET family proteins.Disruption of this tightly regulated methylation–demethylation balance contributes to aberrant bone remodeling, reduced bone mineral density, and increased susceptibility to osteoporosis.
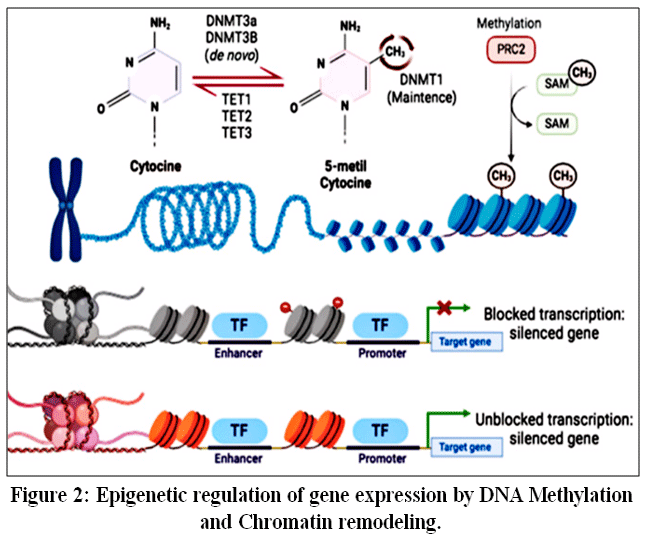 |
Figure 2: Epigenetic regulation of gene expression by DNA Methylation and Chromatin remodeling.
|
Expression of genes is regulated epigenetically through the addition of a methyl group to the carbon 5 of the cytosine. Methylation is performed by the DNMT and TET enzymatic activities, with DNMT adding a methyl group and TET removing methyl groups. The addition of methyl groups causes a condensation of the chromatin, creating a denser form of the chromatin. Due to the density created by the addition of methyl groups, RNA polymerase cannot access the region of the gene for transcription.
Histone modification
Histone modification has significant effects when determining the cell fate of bone cells. The architectural modification of chromatin and its accessibility for transcription through histone modifications has an influence on the expression of osteogenic genes, and is affected by histone acetylation. On the contrary, histone deacetylases (HDAC) promote compaction of chromatin by removing acetylation modifications and hence inhibit osteoblast differentiation by inhibiting RUNX2-mediated transcriptional programs. There are other histone addition (methylation) marks that have distinct regulatory effects; those histone marks which are repressive (H3K27me3) decrease the expression of osteogenic genes, and those that activate (H3K4me3) promote osteoblast maturation and bone formation. There are multiple derangements of histone-modifying enzyme activity under disease circumstances including oxidative stress, glucocorticoid treatment, and metabolic dysfunction, resulting in an imbalance in the metabolic receptor and osteoclast precursors, promoting increased bone resorption and increased skeletal fragility.
The enzymatic action of transferring acetyl groups from acetyl-CoA to lysine residues located on the tails of histones is known as histone acetylation; histone acetylation results in neutralization of the positive charge associated with lysine residues and therefore promotes a relaxed form of the chromatin structure. Acetylation facilitates accessibility of DNA for binding by transcription factors and also recruits proteins containing bromodomains, which further enhance gene activation. Acetyl-CoA is produced through metabolic pathways including glycolysis, fatty acid oxidation, and the tricarboxylic acid cycle; hence, HAT-mediated acetylation utilizes acetyl-CoA as an important substrate, linking metabolic processes in the cell with epigenetic processes. Conversely, deacetylation of histones occurs through the action of HDACs, contributing to condensation of chromatin and suppression of transcriptional activity. Deregulation of the finely-tuned balance between the processes of acetylation, carried out by HATs, and deacetylation, occurring through HDACs, can cause abnormal expression of genes, which may lead to the manifestation of diseases such as cancers that affect bone tissue including osteosarcoma.
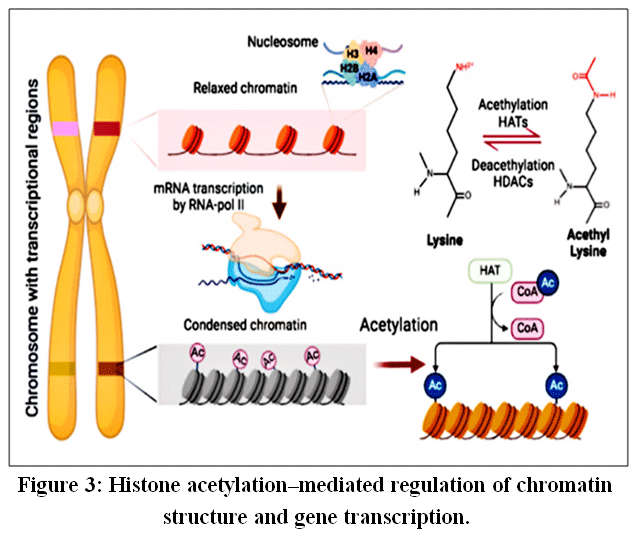 |
Figure 3: Histone acetylation–mediated regulation of chromatin structure and gene transcription.
|
The structural frameworkof DNA is mediated by histone proteins through their ability to create nucleosomes.26 The first10 to 14 amino acids of the N terminal’s histone molecule contain lysine residues capableofbeingacetylated. Theaddition of an acetyl group neutralizes the histone’s positive charge at the lysine residue.27Whenlysinesareneutralized(acetylated),thenucleosomebecomesmorerelaxed, allowing transcription factors and RNA polymerase to haveaneasiertime accessing the gene(s)they will be producing, thus up-regulating transcription. This process is primarily carried out by enzymes known as histone acetyltransferases (HATs), furtherrequiring acetyl-coenzymeAtoperform their work. In contrast to HATs, the enzymes known as histone deacetylases (HDACs) remove the acetyl groups from the lysine amine group, therebycreatingamorecompact (condensed) nucleosome, resulting in transcriptional silencing.28
MicroRNA regulation
MicroRNAs (miRNAs) are small non-coding RNAs (approximately 20–24 nucleotides) that regulate gene expression at the post-transcriptional level and participate in diverse biological processes such as development, cellular differentiation, and disease progression.29 miRNA biogenesis begins in the nucleus, where miRNA genes are transcribed by RNA polymerase II to form long primary miRNA transcripts (pri-miRNAs) containing hairpin structures.30 These pre-miRNAs are processed by the nuclear microprocessor complex composed of DROSHA and its cofactor DGCR8, which cleave them into ~70-nucleotide precursor miRNAs (pre-miRNAs).The pre-miRNAs are subsequently exported to the cytoplasm by Exportin-5 in a Ran-GTP–dependent manner.In the cytoplasm, the RNase III enzyme Dicer further processes pre-miRNAs into a short RNA duplex, from which the passenger strand is degraded while the guide strand is incorporated into the RNA-induced silencing complex (RISC). Mature miRNA within RISC directs gene silencing mainly by inhibiting translation or promoting degradation of target mRNAs.
In bone biology, miRNAs are essential regulators of skeletal homeostasis, influencing osteoblast differentiation, osteoclast activity, and overall bone remodeling. Dysregulation of miRNA expression has been linked to bone-related diseases such as osteoporosis and osteosarcoma.31,32For instance, osteoblast differentiation is positively regulated by miR-29a and negatively regulated by miR-34a, both acting through the TGF-β/Smad signaling pathway.In osteoclasts, miR-21 and miR-155 promote differentiation, whereas miR-26a and miR-223 inhibit osteoclast genesis by modulating RANK/RANKL and MAPK/ERK signaling pathways.Collectively, these observations underscore the critical role of miRNAs in bone remodeling and highlight their potential as therapeutic targets in bone disorders.
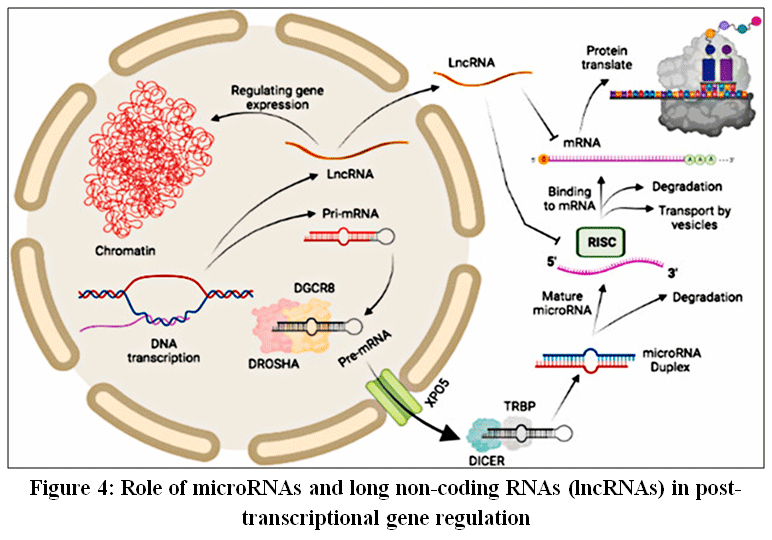 |
Figure 4: Role of microRNAs and long non-coding RNAs (lncRNAs) in post-transcriptional gene regulation
|
DNA encodes microRNAs and lncRNAs that regulate gene translation. MicroRNAs are processed by DROSHA–DGCR8, exported via XPO5, and cleaved by DICER to form mature miRNAs that associate with RISC to suppress mRNA translation. lncRNAs regulate translation by binding mRNAs or sequestering microRNAs, thereby modulating their activity.
Hormonal Influences on Bone Metabolism
Sex Hormones
Estrogen and androgens are key regulators of skeletal homeostasis, acting directly on osteoblasts, osteoclasts, osteocytes, and chondrocytes via estrogen receptors (ERα, ERβ) and androgen receptors (AR). Estrogen suppresses bone resorption primarily by promoting osteoclast apoptosis and reducing RANKL expression while enhancing osteoblast and osteocyte survival and mechanosensing through Wnt/β-catenin signaling. Androgens contribute to periosteal bone expansion and indirectly support trabecular bone maintenance, partly through aromatization to estrogen. Age-related declines in sex steroids—particularly estrogen deficiency after menopause—result in increased oxidative stress, enhanced osteoclast genesis, marrow adiposity, and accelerated bone loss, making sex hormones central to the maintenance of bone mass across the lifespan.33
Parathyroid Hormone (PTH) and PTHrP
Parathyroid hormone (PTH) regulates bone remodelling and calcium homeostasis by binding with receptors found on computer-controlled osteoblasts, osteocytes and bone marrow stroma (bone marrow). PTH promotes osteoblast development directly through Wnt/β-catenin signaling and indirectly through increased RANKL expression, via Gs/cAMP/PKA and PLC/PKC short paths from the PTH1R receptor. The continuous pattern of exposure determines the skeletal response; persistent increases in serum PTH increase bone resorption, while transient administrations of PTH induce anabolic effects. PTHrP has the same actions on osteoclasts as PTH and is necessary for foetal development of endochondral bone and the condition called hypercalcemia.34
Growth Hormone–IGF-1 Axis
Skeletal development and adult bone maintenance rely upon the growth hormone (GH)–insulin-like growth factor-1 (IGF-1) axis. The majority of GH’s anabolic effects on bone are regulated by IGF-1, which is generated both locally and systemically when GH binds to GH receptors on osteoblasts, activating the JAK2/STAT and MAPK pathways. IGF-1 promotes chondrocyte maturation, stabilises Wnt/β-catenin signalling, and promotes osteoblast proliferation, differentiation, and survival. The importance of strictly regulated GH–IGF-1 signalling is highlighted by the fact that while physiological GH/IGF-1 levels encourage bone development, chronic excess, as observed in acromegaly, alters bone microarchitecture and increases fracture risk.35
Leptin
Leptin, that is generated from adipocytes, controls bone metabolism via peripheral and central pathways. Through β2-adrenergic receptors on osteoblasts, leptin acts centrally on hypothalamic nuclei to stimulate sympathetic nervous system activity, resulting in the release of norepinephrine that inhibits osteoblast growth and promotes osteoclast genesis. Peripherally, leptin promotes osteogenesis and chondrogenesis by directly binding to leptin receptors on osteoblasts and bone marrow mesenchymal stem cells. The balance between these conflicting routes, bone type, and physiological setting determines the net skeletal effect of leptin, which explains the inconsistent results shown in both experimental and clinical investigations.36
Adiponectin
Through central and peripheral signalling pathways, adiponectin, which is primarily produced by bone marrow adipose tissue, affects bone remodelling. Adiponectin supports osteoblast survival, mineralisation, and the inhibition of osteoclast genesis under normal settings by peripherally interacting with AdipoR1 and AdipoR2 on osteoblasts to activate AMPK and PI3K signalling. Adiponectin acts on brainstem neurones to decrease sympathetic tone centrally, which indirectly increases bone mass. However, increased adiponectin levels in inflammatory and ageing circumstances may cause signalling to shift in favour of bone resorption, highlighting its context-dependent function in skeletal homeostasis.37
Emerging Techniques in Bone Density Assessment
For the diagnosis of osteoporosis, dual-energy X-ray absorptiometry (DXA) is still the clinical standard for measuring bone mineral density (BMD). However, bone microarchitecture, turnover dynamics, and molecular changes that greatly affect bone strength and fracture risk are not captured by DXA, which mainly evaluates areal BMD. By offering structural, biochemical, genetic, and computational insights into bone quality and skeletal health, emerging technologies seek to overcome these constraints.
High-Resolution Peripheral Quantitative Computed Tomography (HR-pQCT)
Using high-resolution peripheral quantitative computed tomography (HR-pQCT), clinicians and researchers can assess the external appearance of bones in three dimensions. The most common sites of testing with the HR-pQCT are the tibia and radius. HR-pQCT enables researchers to create high-resolution (volumetric) images of both trabecular and cortical compartments, whereas dual-energy X-ray absorptiometry (DXA) uses two-dimensional projections. As a result, the HR-pQCT can provide quantitative measurements of cortical thickness, cortical porosity, trabecular number, trabecular thickness, and trabecular separation, thus providing greater detail on the structure and quality of bone than BMD alone. Research has shown that this assessment tool can be valuable for identifying microarchitectural degeneration that cannot be detected by using only a standard BMD measurement in a research laboratory or specialized practice setting.38
HR-pQCT has proven valuable in differentiating skeletal changes associated with aging, metabolic bone diseases, endocrine disorders, and genetic skeletal conditions. It also enables longitudinal monitoring of bone structural changes in response to pharmacological interventions, making it a powerful adjunct to conventional imaging modalities.
Bone Turnover Biomarkers
Biochemical markers released into the bloodstream during bone formation and resorption are referred to as bone turnover biomarkers, or BTMs. BTMs depict the current rate of skeletal remodelling, in contrast to imaging methods that display cumulative bone mass.
Procollagen type I N-terminal propeptide (P1NP), C-terminal telopeptide of type I collagen (CTX), and osteocalcin tend to be detected blood indicators. While CTX signifies bone resorption activity, P1NP is frequently utilised as a measure of bone production. Bone matrix formation and osteoblast activity are linked to osteocalcin. Since changes in their levels can be identified before changes in bone density, these biomarkers are very effective in tracking therapy response.BTMs are being utilised more often in clinical and research settings to monitor the efficacy of anti-resorptive or anabolic medications, measure fracture risk, and assess patient adherence to therapy. However, biological variability, circadian cycles, and assay standardisation must be carefully taken into account while interpreting them.39
Epigenetic Biomarker Panels
A rapidly emerging field in bone research is epigenetic biomarkers, which give details regarding regulatory modifications that impact skeletal health without changing DNA sequence.40Histone alterations, DNA methylation patterns, and circulating non-coding RNAs, mainly microRNAs, are examples of these biomarkers. Variations in bone density and osteoporosis susceptibility have been correlated with DNA methylation profiles linked to genes related to bones.41 In a similar vein, certain circulating microRNAs have been found to be associated with age-related skeletal degeneration, fracture risk, and bone loss.42These indicators provide a less invasive method for early risk assessment because they can be found in peripheral blood.Currently being investigated as potential methods of predicting when bone loss occurs in patients prior to any visual indicators seen on imaging, epigenetic biomarker panels have mostly yet to be validated outside of research environments, but show promise for potential use in clinical environments in the future.
AI-Based Bone Imaging
Bone imaging analysis is increasingly being enhanced by integrating machine learning and Artificial Intelligence (AI) as part of its analysis process. The use of these technologies improves the ability to acquire additional information from existing imaging modalities such as CT and DXA to provide more precise diagnoses.AI algorithms recognize subtle differences in bone texture, shape, and density distribution, allowing for the identification of subtle patterns in bone that may not otherwise be evident using traditional analytical techniques.43 Automated methods that are able to evaluate the state of an individual’s bone health, quantify the likelihood of fracture, and enhance repeatability by reducing the variability of different observers have also been developed through AI technology.44 AI technology also allows for “Opportunistic Screening,” which involves collecting bone health information from imaging performed for other diagnostic reasons. This approach presents an opportunity to provide more individuals with the ability to assess their risk of fracture and identify early Osteoporosis without increasing their exposure to radiation.45,46
Multi-Omics Approaches
To provide an extensive overview of bone health, multi-omics approaches integrate data from several biological domains, such as transcriptomics, proteomics, metabolomics, genomics, and epigenomics. Multi-omics captures the intricate interactions between genetic predisposition, metabolic status, protein expression, and environmental factors rather than relying on a single measurement. Numerous genetic variations linked to bone density and fracture susceptibility have been found through genomic investigations.47 Additional circulating compounds associated with mineral metabolism and bone turnoverwere identified by proteomic and metabolomic analysis.48These datasets can be integrated to stratify people according to their molecular risk profiles.49Multi-omics techniques are a significant step towards precision medicine in osteoporosis therapy and personalised bone health management, even though they are currently restricted to research and high-resource settings.50
Therapies and Future Directions
Use of personalised and precision-based approaches for the treatment and control of osteoporosis has grown dramatically in recent years. The development of new therapies that involve the use of CRISPR technology to identify and correct high-risk genetic mutations associated with osteopenia and to utilize the transplantation of osteoblasts derived from stem cells to enhance the process of osseointegration has made it possible for people living with osteoporosis to obtain these types of therapies, as well as many others. With respect to epigenetics, it has become clear that the potential exists for therapies aimed at restoring the expression of osteogenic genes through the targeting of DNA methylation/histone modifications, as well as the potential for microRNA therapies to alter the ratio of osteoblasts to osteoclasts to promote the generation of normal bone density and strength.
In addition, the development and availability of various next-generation selective estrogen receptor modulators (SERMS), transdermal hormone delivery systems (i.e., skin patches), and personalised menopausal hormone therapyspecifically on an individualised risk profilerepresents a significant progression in the development and use of hormone-modulating strategies that are both safe as well as tailored to the individual. Smart wearable devices and digital health technology that integrate artificial intelligence-driven analytic capabilities and individual patient health information has led to the creation of new devices that can monitor calcium/vitamin D metabolism and calculate an individual’s risk for fracture (real time) continuously and serve as valuable adjuncts to existing biologically based therapies.
Conclusion
Genetics, epigenetic modifications and hormonal changes, combined with mechanical forces acting on the osteochondral unit, interact with one another in a dynamic fashion to produce the development of osteoarthritis (OA), which is a very complex and multidimensional process. Evidence from this review points to OA being a disease of the entire joint; it includes inflammation of the synovium, remodelling of the subchondral bone, and degradation of the cartilage. Epigenetic changes provide a method for the influence of environmental, metabolic and inflammatory signals to affect the way genes are expressed in the tissue (joint). Additionally, many genetic variants associated with the cartilage matrix proteins, the signalling pathways of the osteochondral unit and bone remodelling regulators create a lower barrier for tissue injury and also result in impaired repair mechanisms for cartilage. The variation seen in the susceptibility of OA development and progression between genders and age groups is due in part to the effects of hormonal regulation on the homeostasis of bone and cartilage.
Technological advances in the evaluation of bone density and quality, including high-resolution imaging, markers of bone remodelling, and combined epigenetic and multi-omics approaches, offer new opportunities to detect early structural and molecular changes which may occur prior to the point when joint becomes irreparably degenerated. These approaches also provide a method for detecting OA at an earlier point in time and for determining the risk of OA and for designing targeted treatments for an individual patient at risk for the disease.
Future studies should focus on translating biologically complex mechanisms of Osteoarthritis into clinically relevant biomarkers & treatments that alter both the cartilage & underlying bone structures of the joint instead of the end stage of the osteoarthritic process.
Acknowledgement
The authors would like to thank Joginpally B.R. Pharmacy College for providing institutional support and facilities.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable.
Author Contributions
- Muthukkuruppan Menaka: Conceptualization, supervision, critical revision of the manuscript.
- Annapareddy Bindusha:Manuscript formatting and technical editing.
- Boyapally Maheshwari Reddy:Literature search, data curation, drafting of the manuscript.
References
- Loeser RF, Goldring SR, Scanzello CR, et al. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697-1707. doi:10.1002/art.34453
CrossRef - Zhu X, Chen T, Dong J, Ruan B, et al. Targeting epigenetic modifiers in osteoarthritis: from biological insights to preclinical practice. Epigenetics Insights. 2025;18:e012. doi:10.1177/25168657251301234
CrossRef - Young DA, Barter MJ, Soul J. Osteoarthritis year in review: genetics, genomics, epigenetics. Osteoarthritis Cartilage. 2022;30(2):216-225. doi:10.1016/j.joca.2021.11.002
CrossRef - Gill R, Liu M, Sun G. Genomic heterozygosity is associated with a lower risk of osteoarthritis. BMC Genomics. 2024;25:85. doi:10.1186/s12864-024-10052-0
CrossRef - Gong Y, Zhu W, Zhu M. Identification and functional characteristics of a novel splicing heterozygote variant of COL2A1 associated with Stickler syndrome type I. Front Genet. 2024;15:1308737. doi:10.3389/fgene.2024.1308737
CrossRef - Van Camp L. Hereditary progressive arthro-ophthalmopathy (Stickler syndrome): clinical and genetic features. Mayo Clin Proc. 2010;85(1):53-62. doi:10.4065/mcp.2009.0337
- Zengini E, Finan C, Wilkinson JM. The genetic epidemiological landscape of osteoarthritis from genome-wide association studies. Bone. 2016;93:49-58. doi:10.1016/j.bone.2016.09.008
CrossRef - Delgado-Calle J. Identification of DNA methylation changes associated with disease progression in subchondral bone with site-matched cartilage in knee osteoarthritis. Sci Rep. 2021;11:14. doi:10.1038/s41598-020-80437-0
- Van Meurs JBJ, Uitterlinden AG. The role of genetic variation in osteoarthritis. Nat Rev Rheumatol. 2012;8(2):80-92. doi:10.1038/nrrheum.2011.193
CrossRef - Atasoy Zeybek A, Showel KK, Nagelli CV, Westendorf JJ, Evans CH. The intersection of aging and estrogen in osteoarthritis. npj Womens Health. 2025;3:15. doi:10.1038/s44294-025-00063-1
CrossRef - Spector TD, MacGregor AJ. Risk factors for osteoarthritis: genetics. Osteoarthritis Cartilage. 2004;12(Suppl A):S39-S44. doi:10.1016/j.joca.2004.01.008
CrossRef - Valdes AM, Spector TD. Genetic epidemiology of hip and knee osteoarthritis. Nat Rev Rheumatol. 2011;7(1):23-32. doi:10.1038/nrrheum.2010.191
CrossRef - Zengini E, Finan C, Wilkinson JM. The genetic epidemiological landscape of hip and knee osteoarthritis. Bone. 2016;85:2-15. doi:10.1016/j.bone.2016.01.015
CrossRef - Boer CG, Hatzikotoulas K, Southam L. Deciphering osteoarthritis genetics across 826,690 individuals from 9 populations. Cell. 2021;184(24):6003-6015.e14. doi:10.1016/j.cell.2021.09.010
CrossRef - Loughlin J. Genetic contribution to osteoarthritis development: current state of evidence. Curr Opin Rheumatol. 2015;27(3):284-288. doi:10.1097/BOR.0000000000000177
CrossRef - Barat-Houari M, Dumont B, Fabre A. The expanding spectrum of COL2A1 gene variants in 136 patients with a skeletal dysplasia phenotype. Eur J Hum Genet. 2016;24(7):992-1000. doi:10.1038/ejhg.2015.205
CrossRef - Palotie A, Vaisanen P, Ott J. Predisposition to familial osteoarthrosis linked to type II collagen gene. Lancet. 1989;1(8644):924-927. doi:10.1016/S0140-6736(89)92833-4
CrossRef - Stattin EL, Wiklund F, Lindblom K, et al. A missense mutation in the aggrecan C-type lectin domain disrupts extracellular matrix interactions and causes dominant familial osteochondritis dissecans.Am J Hum Genet. 2010;86(2):126-137. doi:10.1016/j.ajhg.2009.12.013
CrossRef - Aubourg G, Rice SJ, Bruce-Wootton P, Loughlin J. Genetics of osteoarthritis. Osteoarthritis Cartilage. 2022;30(5):636-649. doi:10.1016/j.joca.2022.01.009
CrossRef - Jazayeri R, Qoreishi M, Hoseinzadeh H. Investigation of the asporin gene polymorphism as a risk factor for knee osteoarthritis in Iran. Am J Orthop. 2013;42(7):313-316.
- Majava M, Hoornaert KP, Bartholdi D, et al. Heterozygous mutations in the COL11A1 gene and genotype–phenotype correlations in type XI collagenopathies. Am J Med Genet A. 2007;143A(3):258-264. doi:10.1002/ajmg.a.31595
CrossRef - Jaenisch R, Bird A. Epigenetic regulation of gene expression. Nat Genet. 2003;33(Suppl):245-254. doi:10.1038/ng1089
CrossRef - Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet. 2010;11(3):191-203. doi:10.1038/nrg2732
CrossRef - Delgado-Calle J, Riancho JA. The role of DNA methylation in bone biology and osteoporosis. Bone. 2012;51(4):564-573. doi:10.1016/j.Bone.2012.06.003
CrossRef - Reppe S. DNA methylation profiles of bone cells in osteoporosis. Bone. 2017;103:243-250. doi:10.1016/j.Bone.2017.07.011
CrossRef - Li K, Han J, Wang Z. Histone modification-centric regulation in osteogenic differentiation. Cell Death Discov. 2021;7:91. doi:10.1038/s41420-021-00464-0
CrossRef - Fu Q, Jiang T. Role of histone modification in osteoporosis. Front Endocrinol (Lausanne). 2022;13:964103. doi:10.3389/fendo.2022.964103
CrossRef - Wellen KE, Thompson CB. Linking metabolism and histone acetylation dynamics. J Biol Chem. 2012;287(52):43515-43523. doi:10.1074/jbc.R112.407114
CrossRef - Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-297. doi:10.1016/S0092-8674(04)00045-5
- Lee Y. MicroRNA maturation: stepwise processing and regulation. Mol Cell. 2003;12:21-31. doi:10.1016/S1097-2765(03)00243-2
- Chen X. MicroRNA dysregulation in osteoporosis and osteosarcoma. Oncotarget. 2016;7:66221-66236. doi:10.18632/oncotarget.11430
CrossRef - Kapinas K. miRNA regulation of osteogenesis. Front Biosci (Landmark Ed). 2012;17:3174-3186. doi:10.2741/4104
CrossRef - Compston JE. Sex steroids and bone. Physiol Rev. 2001;81(1):419-447. doi:10.1152/physrev.2001.81.1.419
CrossRef - Jilka RL. Molecular and cellular mechanisms of intermittent parathyroid hormone. Bone. 2007;40(6):1434-1446. doi:10.1016/j.Bone.2007.03.017
CrossRef - Giustina A, Mazziotti G, Canalis E. Growth hormone, insulin-like growth factors, and the skeleton. Endocr Rev. 2008;29(5):535-559. doi:10.1210/er.2007-0035
CrossRef - DucyP, Amling M, Takeda S, et.al., Leptin inhibits bone formation through a hypothalamic relay. Cell. 2000;100(2):197-207. doi:10.1016/S0092-8674(00)81558-5
CrossRef - Oshima K. Adiponectin increases bone mass by suppressing osteoclastogenesis. J Bone Miner Res. 2012;27(9):1831-1840. doi:10.1002/jbmr.1640
CrossRef - Boutroy S. In vivo assessment of trabecular bone microarchitecture by high-resolution peripheral quantitative computed tomography. J Clin Endocrinol Metab. 2005;90(12):6508-6515. doi:10.1210/jc.2005-1258
CrossRef - Eastell R, Szulc P. Use of bone turnover markers in postmenopausal osteoporosis. Lancet Diabetes Endocrinol. 2017;5(11):908-923. doi:10.1016/S2213-8587(17)30184-5
CrossRef - Delgado-Calle J, Riancho JA. Epigenetic regulation of bone metabolism. Bone. 2017;96:20-27. doi:10.1016/j.bone.2016.12.025
CrossRef - Caldo D, Massarini E, Rucci M, et al. Epigenetics in knee osteoarthritis: a2020–2023 update systematic review. Life (Basel). 2024;14(2):269. doi:10.3390/life14020269
CrossRef - Jin L, Ma J, Chen Z. Osteoarthritis related epigenetic variations in miRNA expression and DNA methylation. BMC Med Genomics. 2023;16:163. doi:10.1186/s12920-023-01597-6
CrossRef - Ou J, Zhang J, Alswadeh M. Advancing osteoarthritis research: the role of AI in clinical, imaging and omics fields. Bone Res. 2025;13(1):48. doi:10.1038/s41413-025-00423-2
CrossRef - Xie H, Li H. Recent advances in imaging techniques and deep learning applications for early diagnosis of knee osteoarthritis: a narrative review. J Musculoskelet Surg Res. 2025;9:423-431. doi:10.25259/JMSR_209_2025
CrossRef - Subramanian B, Kumarasami N, Shastry P. AI-driven pathology detection and osteoarthritis grading from knee radiographs: a multi-site study. Osteoarthritis Cartilage. 2024;32(6):789-798. doi:10.1016/j.joca.2024.02.015
CrossRef - Langsetmo L, Peters KW, Burghardt AJ. Artificial intelligence and advanced imaging in osteoporosis assessment. Bone. 2020;134:115266. doi:10.1016/j.bone.2020.115266
CrossRef - Liu Y, Molchanov V, Brass D, Yang T. Recent advances in omics and integration of multi-omics in osteoarthritis research. Arthritis Res Ther. 2025;27:100. doi:10.1186/s13075-025-03563-2
CrossRef - Wei Y, Qian H, Zhang X. Progress in multi-omics studies of osteoarthritis. Biomark Res. 2025;13:26. doi:10.1186/s40364-025-00732-y
CrossRef - Karasik D, Kiel DP. Genomics, epigenomics, and metabolomics in osteoporosis. Nat Rev Endocrinol. 2016;12(3):163–175. doi:10.1038/nrendo.2015.204
CrossRef - Rai MF, Collins KH, Lang A. Insights from transcriptomic, proteomic, and metabolomic studies in osteoarthritis. Osteoarthritis Cartilage. 2024;32(4):385-397. doi:10.1016/j.joca.2023.11.019
CrossRef
Accepted on: 30-03-2026
Second Review by: Dr. Makhabbah Jamilatun
Final Approval by: Dr. Wagih Ghannam








![]()
![]()
