
Alzheimer’s Disease: Molecular Pathophysiology and Current Therapeutic Management
 , Trushali Ajay Mandhare1, Jayesh Suresh Mujumale1, Pooja Suhas Kashid1, Kishor Vasant Otari2
, Trushali Ajay Mandhare1, Jayesh Suresh Mujumale1, Pooja Suhas Kashid1, Kishor Vasant Otari2 DOI : http://dx.doi.org/10.13005/bbra/3523
Download this article as:
![]()
Alzheimer’s disease is a progressive neurodegenerative disease that ultimately shows severe cognitive decline and loss of intellectual function. Due to its widespread occurrence and substantial socioeconomic burden worldwide, it shows a major public health concern in the twenty-first century. This review highlights currently available therapies as well as emerging treatment approaches for the disease. Alzheimer’s disease is the main cause of dementia and affects a large proportion of individuals which are above 85 years. The disorder is mainly marked by progressive decline in memory and reduced cognitive functioning. Key pathological characteristics involve the build-up of amyloid plaques, development of neurofibrillary tau tangles, and decreased acetylcholine concentrations in the brain. Acetylcholinesterase inhibitors are widely used as first-line therapies in the treatment of Alzheimer’s disease and can offer limited improvement in cognitive function, behaviour, and daily activities. Nevertheless, the overall clinical significance of these effects is still debated. Frequently reported side effects include nausea, vomiting, diarrhoea, dizziness, confusion, and abnormalities in heart rhythm. In patients having moderate to severe Alzheimer’s disease, the N-methyl-D-aspartate receptor antagonist memantine is offer limited short-term improvement in cognition, behavioural symptoms, and functional performance. Memantine is often administered along with an acetylcholinesterase inhibitor. Although sometimes well tolerated, debate continues regarding the extent of its clinically meaningful benefit. While acetylcholinesterase inhibitors and NMDA receptor antagonists may slow disease progression, they are unable to completely halt the advancement of Alzheimer’s disease. Among cholinesterase inhibitors, galantamine, rivastigmine, donepezil, and tacrine are the most extensively investigated agents for Alzheimer’s disease therapy.
KEYWORDS:Acetylcholinesterase Inhibitors; Alzheimer's Disease; Donepezil; Galantamine; NMDA Antagonist; Rivastigmine
Introduction
The main cause of dementia in those above 65 globally is Alzheimer’s disease (AD), a progressively worsening neurodegenerative disorder responsible for nearly 50–70% of dementia cases. Deficits in memory, reasoning, direction, understanding, math, learning ability, language, and judgment are among the many brain functions that are affected by this chronic and progressive disease, primarily at the cortical and hippocampal levels.1
Alzheimer’s affects about half of Americans who are eighty-five years of age or older. Females are affected more frequently than males. Some functional reliance is present in patients with mild disease, such as financial difficulties. Patients with intermediate conditions frequently struggle to drive, are more reliant on others, and have trouble taking a shower and going shopping. Motor and balance problems, as well as complete reliance on caregivers, are characteristics of severe illness.2
Four criteria for diagnosing “probable Alzheimer’s” were established at 1984 by the Alzheimer’s Disease and National Institute of Neurological Disorders and Stroke (NINDS) and the Related Disorders Association (ADRDA).3 In addition to cognitive impairments, there are mental symptoms (apathy, delirium, agitation, depression, hallucinations) associated with neurodegeneration of various brain zones and altered neurotransmission. The symptoms severely disrupt the patient’s daily life, and in more difficult situations, they frequently result in hospitalisation since they create a completely dependent position. After the onset of symptoms, life expectancy is roughly ten years.4 According to the fundamental pathophysiology and neuropathology of AD, intracellular Tau neurofibrillary tangles (NFTs) and extracellular amyloid plaques these are the main histopathological lesions of AD. β-amyloid (Aβ) cleavage produces very insoluble and proteolysis-resistant peptide fibrils, which makes the majority of amyloid or senile plaques (SPs).5 The two enzymes γ-secretase and β-secretase (BACE1) sequentially cleave the massive precursor protein amyloid precursor protein (APP) to form Aβ peptides, of which Aβ42, Aβ40, and Aβ38 are the most frequent types. However, if APP is first acted upon and cleaved by α-secretase rather than β-secretase, Aβ is not produced. The “amyloid hypothesis” said that the brain’s creation of Aβ starts a sequence of events that result in the clinical presentation of AD.6 Up until recently, no therapy for Alzheimer’s disease (AD) had shown to improve cognitive function. Neurotransmitter augmentation, mainly focusing on the cholinergic system, was the main focus of early therapy efforts.7 Tacrine, a cholinesterase inhibitor, was shown to enhance cognitive function in AD patients in 1992 as a result of these efforts.” There has been less success with other therapy strategies for improving cognitive function in AD. Vasodilators like hydergine and so-called ‘nootropic’ drugs that improve animal cognition have not been linked to a clinically significant improvement in AD.8
Pharmacologic treatment of AD aims to improve behavioural symptoms, lessen caregiver burden, or stabilise or slow cognitive and functional deterioration. Cholinesterase inhibitors (ChEIs) and memantine, an NMDA receptor antagonist, these are main two medication classes currently authorised for the cure of AD. When treating AD, ChEIs are seen to be the best option. The ChEIs such as tacrine, rivastigmine, donepezil, and galantamine are used in the treatment of AD is reviewed in this article.9 The structural formula of rivastigmine, a cholinesterase inhibitor (ChEI), differs from that of other ChEIs currently on the market. Since their binding to the acetylcholinesterase enzyme (AChE) is dissolved in a matter of minutes, tacrine and donepezil are categorised as short-acting or reversible drugs.10 Assessing how quickly disease is advancing clinically and also shows how severely the patient is already affected might help evaluate the prognosis for a condition like AD, where there is an ongoing neurodegenerative process.11 Much research has been done on the question of which factor—rate of disease advancement vs. disease severity, or “how fast” vs. “how far”—best predicts the course of AD. While both disease severity and rate of progression appear to be predictive of the course of the illness, the pace of progression may be more significant in predicting prognosis than disease severity.12 Compounds that target intracellular neurofibrillary tangles (NFTs) and extracellular amyloid β (Aβ) plaques, the pathological substrate of the disease, are being studied as treatments. This review discusses new possible disease-modifying therapies for AD which are presently undergoing phase I–III trials, as well as current symptomatic treatments.13
In a similar vein, other strategies have been tried to stop tau phosphorylation, such as aggregation, misfolding, and process prevention. Lastly, both tau and amyloid have been treated with “immunotherapy,” the most recently well-known of which is the use of antibodies against amyloid, such as Biogen’s Aducanumab. Nevertheless, a dose that is neither too high to cause adverse effects nor too low to be beneficial has not yet been discovered.14 Limited attention has been given to identifying the most vulnerable neuronal populations, which is still unknown. The mechanism responsible for the disease’s cell selectivity still has to be clarified. After all, a neurodegenerative disease is not always the result of cell damage, such as that caused by a stroke.15 There has not been much focus on identifying the cells that are most susceptible and figuring out what unique characteristics they have that would make them susceptible to the otherwise common malfunctions of cytotoxicity, tau hyperphosphorylation, and aberrant amyloid cleavage.16
Pathophysiology of Alzheimer’s Disease:
Alzheimer’s disease has complicated as well as multifaceted pathogenesis. The build-up of neurofibrillary tangles and amyloid cerebral plaques, which are made up of axonal protein tau that is abnormally insoluble, is a typical pathological characteristic. Cholinergic neuron involvement results in a decrease in synaptic acetylcholine levels.17 Reduction in neurotransmitter levels, disruption of neuronal networks, mitochondrial impairment, oxidative damage, inflammation, ischemia, defective insulin signalling, and abnormal cholesterol metabolism are among the many factors that may contribute to disease pathogenesis. These mechanisms could serve as potential targets for emerging therapeutic approaches.18
Amyloid-Beta (Aβ) Plaque Formation – The amyloid cascade hypothesis is one of the most widely accepted explanations for AD pathogenesis.
Normal Processing of Amyloid Precursor Protein (APP)
APP is a transmembrane protein normally found in neurons and is processed through two pathways:
Non-Amyloidogenic Pathway (Normal)
- APP is cleaved by α-secretase
- Produces soluble APP fragments that support neuronal growth and survival
- No toxic amyloid-beta is formed.
Amyloidogenic Pathway (Pathological)
- APP is first cleaved by β-secretase (BACE1).
- Subsequently cleaved by γ-secretase.
- Results in the production of Aβ peptides, particularly Aβ42, which is highly insoluble and prone to aggregation.
Consequences of Aβ Accumulation
- Formation of soluble oligomers.
- Aggregation into extracellular amyloid plaques.
- Disruption of synaptic communication.
- Induction of oxidative stress.
- Activation of inflammatory responses.
- Neuronal injury and death.
Tau Protein Hyperphosphorylation and Neurofibrillary Tangles
Tau is a microtubule-associated protein responsible for maintaining neuronal structure and axonal transport.
Normal Function of Tau
- Stabilizes microtubules.
- Supports intracellular transport of nutrients and organelles.
Pathological Changes
- Excessive phosphorylation of tau protein occurs.
- Hyperphosphorylated tau detaches from microtubules.
- Microtubules become unstable and collapse.
- Tau aggregates into paired helical filaments.
Formation of Neurofibrillary Tangles (NFTs)
- Intracellular accumulation of abnormal tau proteins.
- Disruption of neuronal transport systems.
- Progressive neuronal dysfunction and death.
Effects – Synaptic degeneration, Impaired communication between neurons, Cognitive decline, Memory impairment.19,20
Prevention
There are several studies which are evaluated some factors that may influence the incidence of Alzheimer’s disease, including food, socioeconomic factors, medical problems, exposure to the environment, and the use of pharmaceuticals and dietary supplements. Many studies have reported an association between these factors and Alzheimer’s disease; however, a direct cause-and-effect relationship has not yet been confirmed, and it remains unclear whether modifying these factors can reduce the risk of developing the disease.21 Treatment of omega-3 fatty acid consumption, hypertension, cognitive engagement, and physical activity are the numerous preventative strategies that show potential. To further evaluate these findings, however, bigger randomised controlled trials are required. As stated in a recent consensus report from the National Institutes of Health State-of-the-Science Conference on the Prevention of Alzheimer’s Disease and Cognitive Decline, there is not enough data to conclude that any dietary supplement, medication, or modifiable factor can lower the risk of Alzheimer’s disease.22
Table 1: Drugs involved in Treatment Alzheimer’s Disease
| Drugs involved in Treatment Alzheimer’s Disease | |||||
| Drug | Entry Dose
(Start Point) |
Dose Escalation Speed | Final Therapeutic Dose | Clinical Identity Tag | Key Limiting Factor |
| Cholinesterase Inhibitors | |||||
| Tacrine | Ten mg × 4/day | Very Slow (~ 4 months) | 30 – 40 mg 4/day | Liver-limited cholinesterase inhibitor | Hepatotoxicity, frequent dosing. |
| Donepezil | Five mg once daily | Fast (~ 1 month) | 10 mg once daily | Simplest daily regimen | Drug- drug interaction |
| Rivastigmine | One and half mg twice daily | Slow (~ 4 months) | 6 mg twice daily | GI-restricted titration | Nausea and vomiting |
| Galantamine | Four mg twice daily | Slow (~ 4 months) | 12 – 16 mg twice daily | Organ-dependent option | Hepatic or renal Contraindication |
| NMDA Receptor antagonist | |||||
| Memantine | Five mg once daily | Rapid (~ 4 weeks) | 10 mg once daily | Moderate–severe disease agent | Stage- specific use only |
Medications
An enzyme which breaks down acetylcholine, which signifies crucial in memory, is reversibly bound and rendered inactive by acetylcholinesterase inhibitors. The only acetylcholinesterase inhibitor that is authorized for therapy at every stage of the illness is donepezil (Aricept).23 Memantine (Namenda), an N-methyl-D-aspartate (NMDA) receptor antagonist approved for managing moderate-to-severe Alzheimer’s disease, is thought to protect neurons from excitatory amino acid–induced neurotoxicity while maintaining the normal physiological functions of glutamate, a neurotransmitter involved in learning and memory processes. The effectiveness of the medications has typically been evaluated in studies using a variety of assessments, including the Alzheimer’s Disease scale.24
Acetylcholinesterase Inhibitors
The acetylcholinesterase inhibitors are used as a first-line treatment for mild as well as moderate Alzheimer’s disease. The majority of systematic reviews and randomized controlled trials have not discovered any appreciable variations in the efficacy of the different acetylcholinesterase inhibitors. These drugs have different adverse impact profiles despite minor differences in their modes of action. Nausea, vomiting, and diarrhoea are the most frequent side effects; neurological and cardiovascular side effects are similar. The dosage given has a direct impact on the frequency of side effects.25
Donepezil, Tacrine, rivastigmine, and galantamine are the drugs that inhibit cholinesterase. Patients who have asthma, gastric ulcers, bradycardia, or heart block should use them cautiously. Studies support the idea that administering cholinesterase inhibitors could improve symptoms like apathy, psychosis, disinhibition, and agitation by regulating the cholinergic transmission in the areas like frontal, temporal, and orbitofrontal. This is because a cholinergic deficit being linked to the appearance of the psychiatric symptoms of AD. The limbic and paralimbic systems function as a bridge between the emotional and cognitive domains, and the nucleus basalis projects to the cerebral cortex.4
They differ mainly in formulations (rivastigmine continuous-release transdermal patch is available) and pharmacokinetic characteristics (donepezil shows significantly overlong half-life than the other medications and it is dosed once in a day), but not in overall efficacy. Meta-analyzes evaluating both global functioning and cognitive performance have proven their clinical improvement in AD. However, these treatments do not alter the long-term progression of the disease, and their overall clinical benefit remains modest, with a mean improvement of 1.37 points on the Mini-Mental State Examination (MMSE) after six months of therapy. The use of ChEIs in very mild AD (i.e., minor cognitive impairment) which is not supported by the data currently available, and in fact, ChEIs may exacerbate cognition at this early clinical stage.26
Examples of the cholinesterase inhibitors-
Tacrine
Rivastigmine
Donepezil
Galantamine
Despite being few, cost-benefit analyses of cholinesterase inhibitors have not revealed any financial advantages. No double-blind, randomized and placebo-controlled studies have demonstrated that the cholinesterase inhibitors postpone admission to residential care. Extrapolating data from short-term trials and less reliable open-label studies provides weak evidence of a delay. Even though cholinesterase inhibitors are presently the cornerstone of treatment for Alzheimer’s disease, the lots of patients do not experience objective and quantifiable benefit. These medications do not change the illness, and it is unclear if they will be profitable.25
Mechanism Of Cholinesterase Inhibitor
Although several therapeutic models have been explored to enhance cholinergic activity and cognitive function in patients having Alzheimer’s disease, cholinesterase inhibition remains the only cholinergic-based strategy that has demonstrated beneficial outcomes in clinical practice. For the treatment of AD, four ChEls received approval thus far in an Asia Europe and North America. These include galantamine, rivastigmine, donepezil, and tacrine (rarely used due to hepatotoxicity).27 The ChEls preserve certain cholinergic system function while raising ACh levels at synapses and presynaptic receptors. By blocking ChE, which is the enzyme that breaks down ACh in a synaptic cleft, this family of drugs generally increases the availability of ACh. Acetylcholinesterase (AChE) and butyryl-cholinesterase (BuChE) are two ChEs that hydrolytically break down ACh in the brain. ACh is exclusively hydrolysed by AChE, while other choline esters are also hydrolysed by BuChE. Although the exact function of BuChE in humans is unknown, its inhibition may increase the effectiveness of AD treatment.28
Tacrine
Pharmacology of Tacrine
Mechanism and Metabolism: Tacrine reversibly inhibits cholinesterase. The half-life of tacrine is two to four hours and is processed in the liver by the cytochrome P450 1A2 system.
Nausea, vomiting, stomach-aches, anorexia, bradycardia, myalgias, ataxia, and an elevation in liver enzymes (particularly glutamic-oxalacetic transaminase) in 40% of cases are the most notable side effects.29
Use and Monitoring: Patients with hepatic impairment cannot take it. Hepatic enzyme elevation is more common in women and typically happens during the first 12 weeks of treatment. After quitting medication, enzyme levels return to normal in four to six weeks. Enzyme controls must be performed every week for the first six weeks, then every month for the next two months, and finally every quarter after that.
Every six weeks, the dosage must be increased by 40 mg. Velnacrine, suronacrine, and methoxitacrine are derivatives that are being studied. Nearly 90% of patients tolerated it.4
Rivastigmine
Pathology: Cholinergic neural circuits that are essential for learning, memory, and attention are damaged in AD. Acetylcholine (ACh) concentrations drop as a result.
Mechanism: ACh is metabolized at synapses by the enzyme cholinesterase (ChE).
Treatment Action: By blocking this enzyme, ChE inhibitors raise the brain’s supply of ACh.
Result: In AD patients, who’s natural ACh levels are much lower than normal, this elevated ACh aids in enhancing memory and cognitive functioning.
Mechanism of Action: ChEIs raise the amount of acetylcholine in the brain by blocking the enzyme which deactivates it in the synaptic cleft.
Treatment for Symptoms: It should be mentioned that ChEIs target symptoms rather than causes of AD.
Effects of Disease Progression: As AD worsens, butyrylcholinesterase levels rise, and acetylcholinesterase levels fall, with butyrylcholinesterase assuming the role of metabolising acetylcholine.
Rivastigmine Specificity: By covalently attaching toward the active centers of acetylcholinesterase and butyrylcholinesterase., rivastigmine, in contrast to its rivals, selectively inhibits both enzymes.
Metabolism: Rivastigmine is not metabolized in the liver; instead, the breakage of these covalent bonds is the first stage in its breakdown.
Rivastigmine’s pharmacokinetics-
Absorption: After oral administration, absorption is rapid (above 90%), with meal consumption lowering concentration by 30%.
Distribution: Red blood cells are linked to 40–50% of the low protein binding (around 40%).
Metabolism and Elimination: N-demethylated in the liver after being converted to ZNS 144-666 in the central nervous system. Complete removal occurs within 24 hours, with an elimination half-life of less than two hours.
CSF Concentration: Having a half-life ranging from 0.31 to 0.95 hours, the concentration of cerebrospinal fluid rapidly declines.
Dose-dependent Inhibition: The inhibition of the AChE and BuChE enzymes in the CSF was shown to be clearly dose-dependent.
Adverse effects: Nausea, vomiting, anorexia, weight loss, dyspepsia and asthenia were the most frequent adverse effects of rivastigmine reported by participants in clinical trials. Early in the course of the medication, these side effects were more common. As a result, it is advised to take the medication with food. Over time, side symptoms usually go away.30
Donepezil
Pharmacology of Donepezil
Pharmacodynamics
Acetylcholinesterase (AChE) is inhibited by donepezil through a combination of competitive and non-competitive mechanisms.
Selectivity: Compared to butyrylcholinesterase (BuChE), it shows notable selectivity for AChE, which is thought to lead to a decreased frequency of peripheral cholinergic adverse effects.
Comparing this inhibitor to others: Galantamine’s additional allosteric regulation of nicotinic receptors is mentioned, and donepezil’s profile is contrasted with that of galantamine and rivastigmine, pointing out variations in their affinities for AChE and BuChE.
Therapeutic Relevance: At some dosages (5 mg and 10 mg), erythrocyte AChE inhibition is observed; however, the therapeutic significance of the pharmacological variations among inhibitors is still unknown.
Efficacy: In AD patients, donepezil’s efficacy is roughly 64% at five mg and 78% at doses less than ten mg.
Pharmacological Characteristics: The affinity of cholinesterase inhibitors for AChE and BuChE varies: Galantamine and donepezil are selected for AChE than BuChE (50-fold and 1000-fold, respectively).
Pharmacokinetic-Metabolism: The cytochrome P450 isoenzymes, 2D6 and 3A4 break down donepezil inside the liver.
Interactions: Inhibitors of these enzymes, including cimetidine, ketoconazole, paroxetine, fluoxetine, and fluvoxamine, may interact with the medication.
Impact on concentration: It has been observed that ketoconazole and cimetidine raise donepezil plasma concentrations.
FDA recommendations: These observed effects were deemed insignificant under the most recent US FDA recommendations.
Drug Interactions: In healthy individuals, donepezil doesn’t significantly affect the pharmacokinetics of digoxin, theophylline, or warfarin; howevere little changes may be clinically significant.
Metabolism and Excretion: Hepatic first-pass metabolism of donepezil is quite extensive. The majority of metabolites, such as donepezil-cis-N-oxide, 5-O-desmethyl donepezil and 6-O-desmethyl donepezil are identified in the urine, with 11–17% being eliminated unaltered.
Age-Related Variations: Pharmacokinetic characteristics vary by age. Donepezil shows higher apparent volume of distribution and a longer Tmax and half-life in older volunteers than in younger ones.9
Safety Profile: Donepezil therapy was not associated with any clinically meaningful changes in vital signs, haematological parameters, or biochemical investigations, including hepatic, renal, metabolic, electrolyte, creatine phosphokinase, and urinalysis findings.
Tolerability and Side Effects:
In general, donepezil is well tolerated. As is common with cholinesterase inhibitors, nausea, vomiting, and diarrhoea were the most common adverse effect (AE) noted in clinical trials.
The total rate of adverse events is dose-related.
The incidence of AE can be lowered to a rate comparable to a placebo by delaying titration to the 10 mg dose.31
Galantamine
Chemical Formula and Source
Galantamine (C17H21NO3), a tertiary alkaloid that is naturally collected and extracted from plants like the Caucasian snowdrop (Galanthus nivalis), having a molecular mass of a 287.35 g/mol. Synthesis: Techniques for total synthesis have been developed because of the high cost of extraction and the scarcity of natural sources. characteristics: It contains three chiral centers, is mildly lipophilic, and has alkalinic characteristics (pKa: 8.2). The naturally occurring form is S, R, S.32
Mechanism of action
Reduced cholinergic neurons in AD are linked to cognitive impairments like poor learning and memory. Acetylcholine (ACh), a neurotransmitter generated in cholinergic neurons for release into the synaptic cleft, is more readily available in the synaptic cleft when the enzyme AChE is inhibited. This compensates for the decrease in cholinergic function brought on by cholinergic neuron degeneration.31
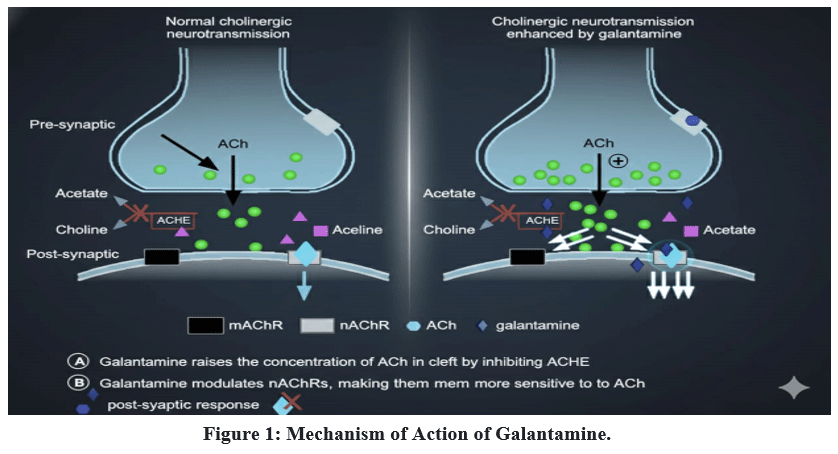 |
Figure 1: Mechanism of Action of Galantamine.
|
Galantamine binds competitively and also reversibly to the active site of AChE to prevent the breakdown of ACh. Galantamine shows its inhibitory effects on AChE in the hippocampus and frontal cortex regions of the brain, these are the two brain regions where cholinergic neurotransmission is most compromised in AD patients, are particularly significant. In vitro, galantamine inhibits AChE 53 times more than butyrylcholinesterase (BuChE) (IC50 values were 0.35 and 18.6 µmolL, respectively). After thirty minutes of a single oral administration of ten mg of galantamine, the median maximum inhibition of red blood cell AChE in healthy volunteers is around 40%. When galantamine (5–15 mg t.i.d.) was administered to AD patients for two to three months, AChE inhibition varied from roughly 20 to 40%.33
When galantamine therapy was stopped, this inhibition quickly decreased, and 30 hours after the last dosage, almost no inhibition was seen. Galantamine may have a tolerability benefit due to its low affinity for BuChE. Although BuChE, which is mostly present in plasma, is not believed to be directly responsible for the cholinergic disruption observed in early AD, its suppression may hurt the periphery.34
Metabolism and pharmacokinetics
Administration: For clinical use, only oral administration is permitted.
Absorption: Following oral consumption, there is high bioavailability (85–100%).
After a single oral dosage, peak plasma concentration (Tmax) happens 0.5 to 2 hours later. Compared to healthy adults, AD patients have Cmax values that are 30–40% higher.
Effect of Food: Consumption of food decreases. increases Tmax by 1.5 hours and Cmax by 25%, CSF Levels: After three months, CSF galantamine levels in patients receiving 16–24 mg/day are about 70 mg/ml; however, after a year, they dramatically drop by about 40%, while plasma levels stay the same.
Protein Binding: Patients taking high dosages of galantamine may experience an increase in its modest plasma protein binding (18–25%).
Elimination: The half-life of plasma elimination is five to seven hours. Renal clearance makes up only 25% of the overall clearance, which is roughly 0.34 l/h·kg.
Metabolism: The enzymes CYP2D6 and CYP3A4 are primarily responsible for the drug’s hepatic metabolism (>75%).
Variations in Metabolizers: Compared to weak metabolizers, extensive CYP2D6 metabolizers generate noticeably more O-demethylated metabolites. Higher amounts of unaltered galantamine and its N-oxide are seen in poor metabolizers.
Clinical Relevance: Although medication clearance is slightly lower in females and poor CYP2D6 metabolizers (~25%), these differences are not thought to be clinically significant enough to call for dosage modifications.
Side effects: Hypertension, asthenia, fever, depression and malaise are among the adverse events (AEs) from clinical trials and post-marketing experience that have been documented in individuals receiving galantamine.35
Efficacy of Cholinesterase-Inhibitor Drugs-
The only medications authorized for Alzheimer’s disease in the US are donepezil and tacrine, must be considered palliative therapies. When compared to a placebo or no treatment, they produce modest but quantifiable improvements in cognitive test scores. Although the advantages of the various cholinesterase inhibitors are the same, the side effects of the medications vary significantly. Tacrine and donepezil offer comparable benefits above placebo at maximal doses, although donepezil can be administered once daily and has fewer side effects.36 Cholinesterase inhibitors are always effective for patients having mild to moderate illness; thus, treatment can start at any point after diagnosis. There is not enough data to suggest that residents of nursing homes receive them. Only uncontrolled extension trials with tacrine, donepezil, and rivastigmine provide evidence in favour of long-term use of cholinesterase inhibitors.37 It is unclear if long-term use of these medications causes tolerance. Gradual increases to the maximum permissible dose have been advised because greater doses offer the greatest benefits and the most negative effects. Although treatment can be continued indefinitely, it is frequently stopped due to a lack of effectiveness or a declining tolerance to adverse effects. When treatment is stopped, patients and their families should be informed that the patient’s condition may worsen.38
N-methyl-D-aspartate antagonist-
Memantine is an additional treatment option for moderate as well as severe AD. This medication is a moderately affinity, non-competitive N-methyl-D-aspartate (NMDA) antagonist that thought to shield neurons from excitotoxicity. After six months of usage, a systematic evaluation of double-blind, parallel-group RCT studies of memantine revealed improvements in behaviours, ADLs, and cognition in individuals with moderate to severe AD.39 Memantine may lessen the psychological and behavioural symptoms of dementia, according to another systematic review that included six RCT studies. In memantine trials, headaches, disorientation, and dizziness were the most common side effects. Agitation may occur in a small percentage of patients.13
Memantine
A non-competitive NMDA antagonist is memantine. In addition to being an agonist of the AMPA receptor, it inhibits the calcium channels of such receptors, preventing calcium from entering neurons and causing damage. Both behaviour and cognition have improved in double-blind studies. In the US, it is now undergoing phase III research. Vomiting, restlessness, vertigo, fatigue, and dizziness are the side effects. In the US, it is now undergoing phase III research.4
Excessive glutamatergic activity is prevented by memantine. The 20 mg daily dose of Memantine is given for 6 months which is benefited patients with up to mild to moderate memory loss by one point on the ADAS-cog, according to Cochrane review, although it is unlikely to have therapeutic relevance. Patients on memantine showed a slight decrease in agitation. To avoid one agitation episode, 17 patients having severe Alzheimer’s disease would require six months of treatment. According to a meta-analysis, memantine had mixed benefits for people with intermediate Alzheimer’s disease. It was unsuccessful for those with mild Alzheimer’s.40 Memantine is always consumed in conjunction with acetylcholinesterase inhibitors and it is generally tolerated. One research shows, patients with severe Alzheimer’s disease on donepezil was randomly assigned to take twenty milligrams of memantine or a placebo daily for twenty-four weeks. Patients treated with memantine showed slight improvements in cognitive function and activities of daily living, with better outcomes reported on the SIB and ADCS-ADL scales compared with placebo-treated patients.41 Another trial assessed the safety as well as efficacy of 20 mg of memantine daily for twenty-four weeks in patients having mild to moderate Alzheimer’s disease who were already on galantamine, rivastigmine, or donepezil. When compared to a placebo, adding memantine didn’t show result in a statistically significant improvement. The absence of side effects was in line with results from earlier studies using memantine monotherapy.2
New Treatments
Recent years (2023-2026) have brought major new treatments for Alzheimer disease, especially drugs that actually target the disease process – not just symptoms. Latest treatments that have entered in market.
Disease-modifying drugs
Lecanemab- (Leqembi) (Approved- 2023-2025)
Type- Monoclonal antibody
Mechanism of Action- Humanized IgG1 monoclonal antibody directed against soluble amyloid-β protofibrils and insoluble amyloid plaques. Promotes microglial-mediated clearance of amyloid deposits from the brain. Reduces amyloid burden and slows downstream neurodegenerative processes.
Clinical Benefits
Approved for patients with early Alzheimer’s disease (mild cognitive impairment or mild dementia due to AD). In the Clarity AD trial, reduced cognitive decline by approximately 27% over 18 months compared with placebo. Delays deterioration in memory, cognition, and daily functioning. Demonstrated significant amyloid plaque reduction on PET imaging.
Action- Removes beta-amyloid plaques from brain.42
Donanemab- (Kisunla) (Approved- 2024)
Action- Clears amyloid plaques
Mechanism of Action- Humanized IgG1 monoclonal antibody targeting pyroglutamate-modified amyloid-β (N3pG-Aβ), a form found predominantly in mature plaques. Facilitates removal of established amyloid plaques through immune-mediated clearance.
Clinical Benefits
FDA-approved for early symptomatic Alzheimer’s disease. TRAILBLAZER-ALZ 2 demonstrated slowing of disease progression by over 20–35%, depending on outcome measures. Significant reduction in amyloid plaque burden. Monthly dosing schedule may improve convenience compared with biweekly therapies.
Given as- Monthly IV infusion.43
Earlier drug
Aducanumab (Aduhelm)
Mechanism of Action- Human IgG1 monoclonal antibody that selectively binds aggregated amyloid-β oligomers and fibrils. Promotes microglial clearance of amyloid plaques. Reduces cerebral amyloid burden.
Clinical Benefits
First disease-modifying anti-amyloid therapy approved for Alzheimer’s disease. Produces substantial reductions in brain amyloid plaques. Established proof-of-concept for amyloid-targeting therapies.
First amyloid-targeting drug (2021).44
New drug for symptoms-
Auvelity (New use approved in 2026)
Mechanism of Action- Combination of dextromethorphan (NMDA receptor antagonist and sigma-1 receptor agonist) and bupropion (CYP2D6 inhibitor and norepinephrine-dopamine reuptake inhibitor). Modulates glutamatergic neurotransmission and neuronal signaling. Being investigated for agitation associated with Alzheimer’s disease.
Clinical Benefits
Oral therapy. Demonstrated reductions in agitation and behavioral symptoms in Alzheimer’s patients in clinical studies. Rapid onset of symptom improvement compared with many neuropsychiatric medications.
New indication- Agitation in Alzheimer’s Patients
Works on neurotransmitters (Not amyloid)
Improved symptomatic drugs-
Zunveyl- (benzgalantamine) (Approved -2025)
Improved version of older drug (Galantamine)
Drugs in pipeline (Future market)
Remternetug- next generation amyloid therapy
GLP-1 drugs (like semaglutide)- being studied.45
Conclusion
In conclusion, AD is a prevalent and expensive illness. There are now a number of mildly beneficial symptomatic therapies that are dose-dependent, clinically observable, and repeatable in clinical studies. For the best results, sustained use of these drugs must be enhanced. Maintaining reasonable expectations regarding a neurodegenerative disease and the available symptom-based therapies, as well as customizing doses and titration for each patient to improve tolerance, can help achieve this. The otherwise unstoppable decline in AD may be considerably altered by a number of medications under investigation that have the potential to modify the illness. These therapies will significantly alleviate the disease’s clinical symptoms and produce significant pharmaco-economic advantages, like a decrease in resource consumption. When accessible, costly disease-modifying drugs will present doctors with a variety of ethical and clinical dilemmas regarding to their indications, as well as choices about whether to combine or discontinue other therapies. Future clinical trials will need to take these difficulties into account.
Acknowledgement
The authors would like to acknowledge the Department of Pharmaceutics, Navsahyadri Institute of Pharmacy, for providing the necessary facilities to conduct this review.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable.
Author Contributions:
- Sakshi Sandip Shingade: Conceptualization, literature survey, writing – original draft, and final manuscript preparation
- Trushali Ajay Mandhare: Supervision, critical revision of the manuscript, and academic guidance
- Jayesh Suresh Mujumale: Conceptualization, literature survey, writing – original draft, and final manuscript preparation
- Pooja Suhas Kashid: Supervision, critical revision of the manuscript, and academic guidance
- Kishor Vasant Otari: Overall supervision, administrative support, and final approval of the manuscript.
References
- Folch J, Ettcheto M, Petrov D, et al. Review of the advances in treatment for Alzheimer disease: strategies for combating β-amyloid protein. Neurologia (Engl Ed). 2018;33(1):47-58. doi:10.1016/j.nrl.2015.03.012
CrossRef - Winslow BT, Onysko MK, Stob CM, Hazlewood KA. Treatment of Alzheimer disease. Am Fam Physician. 2011;83(12):1403-1412.
- Jack CR Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):257-262. doi:10.1016/j.jalz.2011.03.004
CrossRef - Desai AK, Grossberg GT. Diagnosis and treatment of Alzheimer’s disease. Neurology. 2005;64(12 Suppl 3):S34-S39. doi:10.1212/WNL.64.12_suppl_3.S34
CrossRef - Hernández-Zimbrón LF, Gorostieta-Salas E, Díaz-Hung ML, Pérez-Garmendia R, Gevorkian G, Quiroz-Mercado H. Beta amyloid peptides: extracellular and intracellular mechanisms of clearance in Alzheimer’s disease. In: Update on Dementia. IntechOpen; 2016.
CrossRef - Xu X. Gamma-secretase catalyzes sequential cleavages of the AbetaPP transmembrane domain. J Alzheimers Dis. 2009;16(2):211-224. doi:10.3233/JAD-2009-0957
CrossRef - Elmaleh DR, Farlow MR, Conti PS, Tompkins RG, Kundakovic L, Tanzi RE. Developing effective Alzheimer’s disease therapies: clinical experience and future directions. J Alzheimers Dis. 2019;71(3):715-732. doi:10.3233/JAD-190507
CrossRef - Aisen PS, Davis KL. The search for disease-modifying treatment for Alzheimer’s disease. Neurology. 1997;48(5 Suppl 6):S35-S41. doi:10.1212/WNL.48.5_suppl_6.35S
CrossRef - Tsuno N. Donepezil in the treatment of patients with Alzheimer’s disease. Expert Rev Neurother. 2009;9(5):591-598. doi:10.1586/ern.09.23
CrossRef - Jann MW. Rivastigmine, a new-generation cholinesterase inhibitor for the treatment of Alzheimer’s disease. Pharmacotherapy. 2000;20(1):1-12. doi:10.1592/phco.20.1.1.34664
CrossRef - Shin D, Lee S, Kim JP, et al. Biomarker-integrated prognostic stagings for Alzheimer’s disease. Nat Commun. 2026;17(1):2235. Published February 2, 2026. doi:10.1038/s41467-026-68732-6
CrossRef - Farlow MR, Hake A, Messina J, Hartman R, Veach J, Anand R. Response of patients with Alzheimer disease to rivastigmine treatment is predicted by the rate of disease progression. Arch Neurol. 2001;58(3):417-422. doi:10.1001/archneur.58.3.417
CrossRef - Yiannopoulou KG, Papageorgiou SG. Current and future treatments for Alzheimer’s disease. Ther Adv Neurol Disord. 2013;6(1):19-33. doi:10.1177/1756285612461679
CrossRef - Cummings J, Aisen P, Apostolova LG, Atri A, Salloway S, Weiner M. Aducanumab: appropriate use recommendations. J Prev Alzheimers Dis. 2021;8(4):398-410. doi:10.14283/jpad.2021.41
CrossRef - Cecerska-Heryć E, Pękała M, Serwin N, et al. The use of stem cells as a potential treatment method for selected neurodegenerative diseases: review. Cell Mol Neurobiol. 2023;43(6):2643-2673. doi:10.1007/s10571-023-01344-6
CrossRef - Small GW, Greenfield S. Current and future treatments for Alzheimer disease. Am J Geriatr Psychiatry. 2015;23(11):1101-1105. doi:10.1016/j.jagp.2015.08.006
CrossRef - Chen ZR, Huang JB, Yang SL, Hong FF. Role of cholinergic signaling in Alzheimer’s disease. Molecules. 2022;27(6):1816. Published March 10, 2022. doi:10.3390/molecules27061816
CrossRef - Hein ZM, Karikalan B, Gopalakrishna PK, et al. Toward a unified framework in molecular neurobiology of Alzheimer’s disease: revisiting the pathophysiological hypotheses. Mol Neurobiol. 2026;63:282. doi:10.1007/s12035-025-05602-0
CrossRef - Liu E, Zhang Y, Wang JZ. Updates in Alzheimer’s disease: from basic research to diagnosis and therapies. Transl Neurodegener. 2024;13(1):45. doi:10.1186/s40035-024-00432-x.
CrossRef - Sirisi S, Sánchez-Aced E, Belbin O, Lleó A. APP dyshomeostasis in the pathogenesis of Alzheimer’s disease: implications for current drug targets. Alzheimers Res Ther. 2024;16(1):144. doi:10.1186/s13195-024-01504-w.
CrossRef - Mertaş B, Boşgelmez Iİ. The role of genetic, environmental, and dietary factors in Alzheimer’s disease: a narrative review. Int J Mol Sci. 2025;26(3):1222. Published January 30, 2025. doi:10.3390/ijms26031222
CrossRef - Kane RL, Butler M, Fink HA, et al. Interventions to Prevent Age-Related Cognitive Decline, Mild Cognitive Impairment, and Clinical Alzheimer’s-Type Dementia. Agency for Healthcare Research and Quality; 2017.
- Vecchio I, Sorrentino L, Paoletti A, Marra R, Arbitrio M. The state of the art on acetylcholinesterase inhibitors in the treatment of Alzheimer’s disease. J Cent Nerv Syst Dis. 2021;13:11795735211029113. Published July 7, 2021. doi:10.1177/11795735211029113
CrossRef - Terry AV Jr, Callahan PM, Hall B, Webster SJ. Alzheimer’s disease and age-related memory decline (preclinical). Pharmacol Biochem Behav. 2011;99(2):190-210. doi:10.1016/j.pbb.2011.02.002
CrossRef - Waite LM. Treatment for Alzheimer’s disease: has anything changed? Aust Prescr. 2015;38(2):60-63. doi:10.18773/austprescr.2015.018
CrossRef - Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179(2):312-339. doi:10.1016/j.cell.2019.09.001
CrossRef - Balázs N, Bereczki D, Kovács T. Cholinesterase inhibitors and memantine for the treatment of Alzheimer and non-Alzheimer dementias. Ideggyogy Sz. 2021;74(11-12):379-387. doi:10.18071/isz.74.0379
CrossRef - Prešern U, Goličnik M, Bavec A. Genetic and functional dynamics of butyrylcholinesterase in Alzheimer’s disease: from mechanisms to clinical relevance. Chem Biol Interact. 2026;423:111809. doi:10.1016/j.cbi.2025.111809
CrossRef - Nordberg A, Svensson AL. Cholinesterase inhibitors in the treatment of Alzheimer’s disease: a comparison of tolerability and pharmacology. Drug Saf. 1998;19(6):465-480. doi:10.2165/00002018-199819060-00004
CrossRef - Müller T. Rivastigmine in the treatment of patients with Alzheimer’s disease. Neuropsychiatr Dis Treat. 2007;3(2):211-218. doi:10.2147/NDT.S2007.3.2.211
CrossRef - Wilkinson DG. The pharmacology of donepezil: a new treatment of Alzheimer’s disease. Expert Opin Pharmacother. 1999;1(1):121-135. doi:10.1517/14656566.1.1.121
CrossRef - Prvulovic D, Hampel H, Pantel J. Galantamine for Alzheimer’s disease. Expert Opin Drug Metab Toxicol. 2010;6(3):345-354. doi:10.1517/17425251003592137
CrossRef - Robinson DM, Plosker GL. Galantamine extended release in Alzheimer’s disease: profile report. Drugs Aging. 2006;23(10):839-842. doi:10.2165/00002512-200623100-00006
CrossRef - Lilienfeld S. Galantamine—a novel cholinergic drug with a unique dual mode of action for the treatment of patients with Alzheimer’s disease. CNS Drug Rev. 2002;8(2):159-176. doi:10.1111/j.1527-3458.2002.tb00221.x
CrossRef - Razay G, Wilcock GK. Galantamine in Alzheimer’s disease. Expert Rev Neurother. 2008;8(1):9-17. doi:10.1586/14737175.8.1.9
CrossRef - Massoud F, Gauthier S. Update on the pharmacological treatment of Alzheimer’s disease. Curr Neuropharmacol. 2010;8(1):69-80. doi:10.2174/157015910790909520
CrossRef - Havreng-Théry C, Oquendo B, Zolnowski-Kolp V, et al. Cholinesterase inhibitors and memantine are associated with a reduced mortality in nursing home residents with dementia: a longitudinal observational study. Alzheimers Res Ther. 2024;16(1):117. Published May 29, 2024. doi:10.1186/s13195-024-01481-0
CrossRef - Mayeux R, Sano M. Treatment of Alzheimer’s disease. N Engl J Med. 1999;341(22):1670-1679. doi:10.1056/NEJM199911253412207
CrossRef - Olivares D, Deshpande VK, Shi Y, et al. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer’s disease, vascular dementia and Parkinson’s disease. Curr Alzheimer Res. 2012;9(6):746-758. doi:10.2174/156720512801322564
CrossRef - Matsunaga S, Kishi T, Iwata N. Memantine monotherapy for Alzheimer’s disease: a systematic review and meta-analysis. PLoS One. 2015;10(4): e0123289. Published April 10, 2015. doi:10.1371/journal.pone.0123289
CrossRef - Atri A, Molinuevo JL, Lemming O, Wirth Y, Pulte I, Wilkinson D. Memantine in patients with Alzheimer’s disease receiving donepezil: new analyses of efficacy and safety for combination therapy. Alzheimers Res Ther. 2013;5(1):6. Published January 21, 2013. doi:10.1186/alzrt160
CrossRef - van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388(1):9-21. doi:10.1056/NEJMoa2212948
CrossRef - Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer’s disease. N Engl J Med. 2021;384(18):1691-1704. doi:10.1056/NEJMoa2100708
CrossRef - Knopman DS, Jones DT, Greicius MD. Failure to demonstrate efficacy of aducanumab: an analysis. Nat Rev Neurol. 2021;17(6):369-370. doi:10.1038/s41582-021-00515-5
- Cummings J, Lee G, Nahed P, et al. Alzheimer’s disease drug development pipeline: 2024. Alzheimers Dement (N Y). 2024;10(1):e12450. doi:10.1002/trc2.12450
CrossRef
Abbreviations
AD: Alzheimer’s Disease
ADRDA: Alzheimer’s Disease and Related Disorders Association
NINDS: National Institute of Neurological Disorders and Stroke
NFTs: Neurofibrillary Tangles
Aβ: β-amyloid
SPs: Senile Plaques
BACE1: β-secretase
APP: Amyloid Precursor Protein
ChEIs: Cholinesterase inhibitors
NMDA: N-methyl-D-aspartate
AChE: Acetylcholinesterase enzyme
MMSE: Mini-Mental State Examination
BuChE: Butyryl-cholinesterase
US FDA: United State Food Drug Administration
Cmax: Maximum plasma concentration
AE: Adverse events
CSF: Cerebrospinal Fluid
Tmax: Time to Maximum Concentration
RCT: Randomised Controlled Trial
ADL: Activities of Daily Living
AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
ADAS-cog: Alzheimer’s Disease Assessment Scale- Cognitive Subscale
SIB: Severe Impairment Battery
ADCS-ADL: Alzheimer’s Disease Cooperative Study- Activities of Daily Living
Accepted on: 13-06-2026
Second Review by: Dr. Sarraa Dhiaa Kasim
Final Approval by: Dr. Eugene A. Silow








![]()
![]()
