
Drug Repurposing against Anhydro-N-acetylmuramic Acid Kinase of Multi-Drug Resistant Acinetobacter baumannii: An in Silico Approach
 and Priya Mondal
and Priya Mondal Department of Microbiology, The University of Burdwan, Burdwan, India.
Corresponding Author E-mail:dgupta@microbio.buruniv.ac.in
DOI : http://dx.doi.org/10.13005/bbra/3184
Download this article as:
![]()
Acinetobacter baumannii, a gram-negative coccobacillus is accountable for different nosocomial diseases. It has been enlisted in the ‘critical’ category in WHO published list depending on the urgency for novel drug development as it becomes multidrug resistant (MDR). The aim of this study was to find a drug which can be repurposed against any drug target of these bacteria and thus the time and cost required for typical drug development procedure can be bypassed. In this study, Anhydro-N-acetylmuramic acid kinase (AnmK) of Acinetobacter baumannii was analyzed to be a good drug target which is responsible for the structural integrity of the cell wall of these bacteria. The expression probability of the protein is high with 0.916. PROTPARAM analysis shows that it is a thermostable, non polar protein with molecular weight of 41.7 kDa and pI in the acidic range. The structure prediction was done with SWISS-MODEL (with 46.71% identity with the template) and was found reliable with 91.8% amino acid in allowable region. This predicted structure was used for dug repurposing in which drugs are screened from ZINC15 database (containing FDA approved drug) to find their effective binding (if any) with this protein. PyRx software was used for the docking process which found Ergotamine as the most promising repurposed drug in terms of binding energy(-10.5 kcal/mole) and vina score(-10.3 kcal/mole). Molecular Dynamics Simulation shows that binding of this drug with the protein target is stable over picoseconds time scale.
KEYWORDS:Anhydro-N-acetylmuramic acid kinase; Drug repurposing; MDR; PROTPARAM; PyRx; ZINC15
Introduction
According to a list of pathogenic bacteria published by WHO (2017) there are three reported categories depending on the urgency for novel drug development; Critical, High and Medium. Bacteria enlisted in critical (Priority 1) list are mostly multi drug resistance (MDR) and Acinetobacter baumannii is one of them 1. Acinetobacter baumannii, a gram-negative coccobacillus is involved in health care associated infections (HAIs) to cause nosocomial diseases 2. Acinetobacter baumannii can cause disease of blood, urinary tract, and lungs (pneumonia), or in wounds where its colonization shows significant symptoms, especially in respiratory secretions (sputum) or open wounds 3. The resistant antibiotics can be categorised in first and second line antibiotics. First line antibiotics include Anti pseudomonal cephalosporins (ceftazidime or cefepime), Anti pseudomonal carbapenems (imipenem or meropenem), Ampicillin/sulbactam, Fluoroquinolones (ciprofloxacin or levofloxacin), Aminoglycosides (gentamicin, tobramycin, or amikacin) and Trimethoprim-sulfamethoxazole. Second line antibiotics include Polymyxins (polymyxin B and colistin), Tetracycline derivatives (Minocycline, Doxycycline, Tigecycline) as well as other tetracycline based agent (Eravacycline and Omadacycline). The organism can evolved into drug resistant strains due to accumulation of diverse mechanisms of resistance 4. Certain types of carbapenems (i.e., meropenem, imipenem) are though profoundly bactericidal but defenceless against strain of Acinetobacter 5. The mechanism of action of drug resistance against these drugs is caused by accumulation of multiple antibiotic resistance genes in Acinetobacter species which are capable of developing multidrug-resistant or extensively drug-resistant strains 6, 7. As often as possible cause of MDR mechanisms in nosocomial strains of Acinetobacter baumannii is the efflux pumps system; over-expression of bacterial efflux pumps can decrease the concentration of beta- lactam antibiotics in the peri-plasmic space. Behind clinical resistance seen in Acinetobacter, efflux pumps as a rule act in affiliation with over expression of AmpC beta-lactamases or carbapenems. Efflux pumps can evacuate beta-lactam antibiotics as well as quinolones, tetracyclines, chloramphenicol, and tigecycline 8. These pumps cause the leakage of antibiotics and leaked out substances from the bacteria, creating multidrug resistance 9.
Drug repurposing is the method of recognising novel therapeutic prospects of existing drugs and finding treatments for untreated infections 10. In case of drug development, the total procedure takes lots of time for the preclinical and clinical trial phase and also needs a profound investigation for its harmful side effects (if any) before its application on human. But in case of drug repurposing, the total procedure is time and cost effective. As in this method the drug is searched from list of preapproved drug, the method of the preclinical and clinical trials can be bypassed and using these drugs will have minimum human risk. In this work, a set of steps were followed for the selection of a drug target which proposed Anhydro-N-acetylmuramic acid kinase as one of the drug target and then drug repurposing method was applied for searching of drug from FDA approved drug list.
Materials and Method
Selection of protein for using as drug target
The Integrated Microbial Genomes (IMG) v.5.0 site (https://img.jgi.doe.gov/) was used to find proteins in Acinetobacter baumannii ATCC 17978 which have homologues in other Acinetobacter baumannii ATCC genomes but absent in Human for selection of functional protein that can be used as probable drug targets. The parameters of the said searching methods are kept as default. For cross checking the absence of the selected protein in Homo sapiens, it was again searched with BLASTp v.2.2.17 with human taking the FASTA sequence of this protein (which was downloaded and saved in doc file) 11, 12.
Exploring different parameters of the selected protein
The different physicochemical parameters of the selected protein were retrieved through Expasy PROTPARAM 13.
Finding the localization of selected protein
Determining the localization of selected protein was done by PSORTb v.3.0 software (https://www.psort.org/psortb/) and cross-checked by SCLpred-EMS 14, 15.
Finding expression probability of the selected protein
CAIcal (Codon Adaptation Index calculator) (http://genomes.urv.es/CAIcal/) 16 was used to find the expression probability of the selected protein (through its gene sequence) which is important for any functional protein.
Checking the presence of protein in genera other than Acinetobacter:
For finding the presence of the selected protein in bacterial species other than Acinetobacter, it was searched through BLAST excluding the Acinetobacter species (taking 60% as the cut off of identity). Taking the protein sequences from the selected genera along with the protein of choice, Multiple Sequence Alignment (MSA) was performed by Clustal Omega v.1.2.4 (https://www.ebi.ac.uk/Tools/msa/clustalo/) to find the conserved pattern of this protein in different bacterial species 17.
Analysis of protein-protein interaction
Prediction of protein-protein interactions was done by STRING v.11.5 software (https://string-db.org/) 18 which is a knowledgebase software tool.
Predicting & validating the structure of the selected protein
As there is currently no experimentally determined structure of Anhydro-N-acetylmuramic acid kinase(AnmK) Acinetobacter baumannii, the structure of this protein was computationally predicted using two approaches: Homology based and de novo based which were done by SWISS MODEL(https://swissmodel.expasy.org/) and I-TASSER (https://zhanggroup.org/I-TASSER/)server respectively 19, 20. Predicted model structure was visualised by Pymol Molecular Visualization v.2.5 (https://pymol.org/2/) 21 software. SAVES v.6.0 (https://saves.mbi.ucla.edu/) software was used to validate the structure of the predicted protein. SAVES v.6.0 was a multi tool based software 22, 23, 24, 25, 26.
Active site determination
The active site of selected protein was determined using CASTp v.3.0 (http://sts.bioe.uic.edu/), Prankweb v.2.4 (https://prankweb.cz/) and Cbdock2 software 27, 28, 29. Among the predicted output shown by these three softwares, common position with amino acid residues were selected as final active site residues.
Ligand library preparation, docking and its visualisation
A ligand library was generated from FDA approved drugs from the ZINC15 (https://zinc15.docking.org/) database 30 (which consists of 1615 drugs) by retrieving the structure data files (sdf format) of those drugs. Multiple selections of drugs retrieve 100 drugs from the total collection and were chosen for docking with AnmK by PyRx v.1.0 (https://pyrx.sourceforge.io/) software 31. These sdf files were energy minimized with Amber force field and changed over to auto dock compatible file formats (pdbqt) using Open Babel (an inbuilt structural format changing tool of PyRx). Rechecking the docking and the vina score was performed with Cbdock2 software and the docking poses were visualized by Pymol software.
Molecular Dynamics Simulation of the protein and protein-ligand complex
Molecular dynamics simulations of the protein and the complexes (protein-substrate and protein-repurposed drug) were performed using MDweb (https://mmb.irbbarcelona.org/MDWeb/) software 32 which is actually popular windows based MDS software that mimics the algorithm of GROMACS.
Result and Discussion
Selection of protein as drug target
The protein selected using IMG v.5.0 was Anhydro-N-acetylmuramic acid kinase (AnmK) which is present in all strains of the Acinetobacter but not in human. Proteins may be initially considered as a good drug target if it present in all strains of the concerned bacteria but absent in human, Here, for cross checking the absence of AnmK protein in Homo sapiens it was again tested with BLASTp v.2.2.17 with human and was to have no significant similarity in human proteome (Fig. 1). This makes AnmK suitable for being analysed as novel drug target.
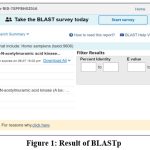 |
Figure 1: Result of BLASTp
|
Different parameters of the selected protein: The resulting values of the parameters of Protparam were appeared in Table 1.
Table 1: Different physicochemical parameters of the selected protein
|
PARAMETERS |
VALUE |
|
Molecular Weight |
41719.23 |
|
pI |
5.24 |
|
Negatively charged residues |
47 |
|
Positively charged residues |
30 |
|
Instability index |
39.61 |
|
Aliphatic index |
90.56 |
|
GRAVY (Grand Average of Hydropathy) |
-0.212 |
The molecular weight of protein is 41.7Kd and its pI value is 5.24. The isoelectric point (pI) is the pH at which a particular molecule carries no net electric charge. The molecular weight and the pI value can be used for effective separation of the protein through SDS PAGE. Numbers of negatively and positively charged residues were found to be 47 and 30 respectively. The instability index provides an estimate of the stability of a protein; a protein with instability index smaller than 40 is anticipated as stable. As the instability index of AnmK protein is predicted to be 39.61, it can be considered as stable. The aliphatic index of a protein is defined as the relative volume occupied by aliphatic side chains (alanine, valine, isoleucine, and leucine). Average aliphatic index of proteins above 80% confirms their thermostability 33. So this protein with aliphatic index value of 90.56 can be considered as thermostable. GRAVY score can be calculated as the sum of the hydropathy values for all the amino acids in a protein divided by the total number of residues in it. Negative GRAVY value indicates that the protein is non-polar and positive value indicates that the protein is polar 34. So, GRAVY value of this protein (-0.212) means the protein is non polar.
Localization of protein
The localization of selected protein (AnmK) was found to be cytoplasmic with a value of 8.96 through PSORTbv.3.0 software. Taking the cumulative localization scores for the 5 sites used by PSORTb, the final prediction was found to be cytoplasmic. Localization was rechecked using SclpredT (It works by EMS response) which predicted its localization as non EMS (Endo Membrane System) that also supports its localization as cytoplasmic region with high confidence level of 8.
Expression probability of the selected protein
The expression probability of the AnmK protein was found with value of 0.916 which is high enough to consider it as a protein with high expression (Fig. 2).
 |
Figure 2: Result of CAIcal server
|
Conservedness of protein
MSA was performed by Clustal Omega v.1.2.4 to know the AnmK protein has conservedness in different bacterial species. Taking 60% as the cut off identity, showed its presence in the genera Klebsiella, Moraxellaceae, Serratia, Gammaproteobacteria, Escherichia, Pseudomonadales, Alkanindinges, Aquirhabdus, Candidatus, Oppiella.
Determination of protein-protein interaction
STRING analysis of the selected protein shows the proteins with which the protein of interest (A1S_0013) interacts (Fig. 3). It shows that the selected protein has high interaction with beta-N-acetyl-D-glucosaminidase (A1S_0492) (0.993) which cleave alternate but chemically equivalent glycosidic bond in the NAG-NAM polymers 35. It has been reported that the enzyme beta-N-acetyl-D-glucosaminidase can be used as drug target; this also supports the probability of AnmK of acting as drug target. The second highest interacting protein is putative phosphoglucose isomerase (0.904) which is involved in glycolysis pathway. That means blocking this protein may have an impact on the glycolysis pathway of the bacteria.
 |
Figure 3: Result of STRING.
|
Predicting & validating the structure of the selected protein
The structure of the protein was predicted through homology modelling using 3qbw1.A as template which has 46.71% identity with the protein of interest. The protein was verified through the SAVES server which shows the overall quality factor found through ERRAT is 89.95 for both the chain A and B. Verify 3D shows that for both the chain, the average score is in the positive range. Though in WHATCHEK some errors and warnings are present, majority functions were passed with success the reflection of which was found in the Ramachandran plot as the presence of 91.8% amino acid in allowable regions (Fig. 6) making the overall structure a stable one.
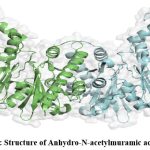 |
Figure 4: Structure of Anhydro-N-acetylmuramic acid kinase
|
 |
Figure 5: Result of SAVES v.6.0
|
 |
Figure 6: Result of PROCHECK
|
Active site determination
Active site prediction was done by submitting the model structure of AnmK in three different active site prediction tools; the common amino acid residues found from these softwares along with their position was given in the following table (Table 2). As the protein of interest (AnmK) is reported to be a homo-dimeric protein, any one chain of a particular subunit can be used for the active site prediction purposes 36. So it can be assumed that this site, which is the site for natural substrate 1,6-anhydro-N-acetylmuramic acid (anhMurNAc) of this enzyme, may be used by the proposed inhibitor (repurposed drug) for binding.
Table 2: Position and name of amino acid residues in active site.
|
POSITION |
AMINO ACID RESIDUE |
|
9 |
MET |
|
11 |
GLY |
|
12 |
THR |
|
94 |
HIS |
|
97 |
THR |
|
100 |
HIS |
|
107 |
THR |
|
109 |
GLN |
|
129 |
ARG |
|
139 |
GLY |
|
140 |
ALA |
|
141 |
PRO |
|
142 |
LEU |
|
143 |
VAL |
|
162 |
ASN |
|
166 |
ILE |
|
168 |
ASN |
|
184 |
ASP |
|
238 |
THR |
|
338 |
GLU |
|
342 |
PHE |
Ligand library preparation, docking and its visualization
As the aim of the study was to find repurposed drug for the selected protein (being used as drug target), the ligand library was prepared from the FDA approved drugs taken from ZINC 15 database. After docking the ligands (approved drugs) with the target protein the ease of interaction was evaluated in terms of binding energy and vina score (in kcal/mol) and compared with that of natural ligand (substrate) of the selected protein which is 1,6-anhydro-N-acetylmuramic acid (anhMurNAc). Depending on the score, a list was prepared taking the best scoring ligands which is given in Table 3. It was shown that the binding energy and the vina score of the natural ligand i.e. the substrate with the protein are -6.7 and -7.3 kcal/mol respectively. The most promising ligand (FDA approved drug) which shows less binding energy and vina score than that of natural ligand is ergotamine with binding energy and vina score of -10.5 kcal and -10.3 kcal respectively. When the docking pose of ergotamine with the protein Anhydro-N-acetylmuramic acid kinase (AnmK) was rechecked with the docking software cbdock2, it was found that the volume of the activity formed by ergotamine is larger (4182 A°3)(Table 4) than that of its normal substrate (1371 A°3).
Table 3: Docking result of selected protein-substrate and protein-drug.
|
PROTEIN NAME
|
LIGAND |
BINDING ENERGY (Kcal/mol) [PyRx] |
VINA SCORE (Kcal/mol) [Cbdock2] |
|
Anhydro-N-acetylmuramic acid kinase (AnmK) |
1,6-anhydro-N-acetylmuramic acid (anhMurNAc) |
-6.7 |
-7.3 |
|
ZINC52955754 (Ergotamine) |
-10.5 |
-10.3 |
|
|
ZINC3816287 (Axitinib) |
-10.5 |
-10.2 |
|
|
ZINC242548690 (Digoxin) |
-10.3 |
-9.4 |
|
|
ZINC3833846 (Nelfinavir) |
-10.2 |
-9.5 |
|
|
ZINC607986 (Nebivolol) |
-9.8 |
-10.1 |
Table 4: Docking parameters of protein-ergotamine as obtained from cbdock2.
|
CURPOCKET ID
|
VINA SCORE |
CAVITY VOLUME(A°3 ) |
|
C1 |
-10.3 |
4182 |
|
C4 |
-9.9 |
561 |
|
C5 |
-9.7 |
551 |
|
C2 |
-8.8 |
3864 |
|
C3 |
-0.6 |
1371 |
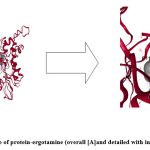 |
Figure 7: Docking pose of protein-ergotamine (overall [A]and detailed with interacting residues [B])
|
Ergotamine is a drug which is used for migraine with the trade name of “Gynergen” 37. Dihydroergotamine has also been proved to act against filarial infection against Wolbachia 5’-aminolevulinic acid synthase 38. The detail of the docking pose of protein-substrate (as positive control) and protein-ergotamine are shown in Fig 7[A] & [B]. It was seen in the picture that there is a difference in the orientation of binding of ergotamine with the protein target than that of its natural substrate and this change may be responsible for its less binding energy.
Molecular Dynamics Simulation of the protein and complexes of protein-ligand and protein-repurposed drug
To check the change in orientation of the protein structure (if any) in isolation as well as upon binding with the substrate and ligand, in course of time, molecular dynamics simulation of the abovementioned structures were performed. This type of simulation is also provides a scope to investigate the deviation of the protein backbone (as RMSD value) and compactness (as radius of gyration) of the protein upon binding with ligand (if any) in nanosecond time scale. The graph of RMSD with amino acid residue shows no drastic variation in the active site region and no change was seen in deviations from the backbone upon binding with the substrate as well as with ligand (repurposed drug) (Fig. 8). So it can be assumed that the binding of the proposed inhibitor of the selected protein (ergotamine) will not hamper its structural stability.
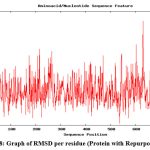 |
Figure 8: Graph of RMSD per residue (Protein with Repurposed drug)
|
 |
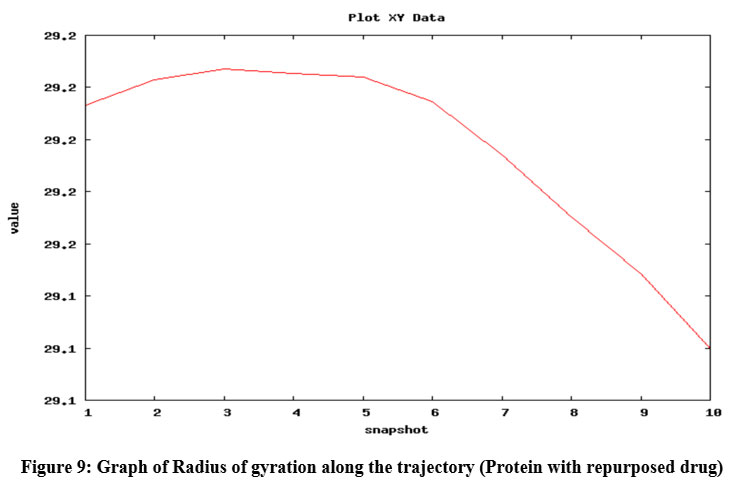
Figure 9: Graph of Radius of gyration along the trajectory (Protein with repurposed drug)
|
Radius of gyration shows the compactness of protein 39; here, radius of gyration of the protein was checked in isolated condition as well as upon binding with ligands (natural substrate and repurposed drug) and it was found that the radius of gyration was not altered upon binding with substrate and repurposed drug (Fig. 9) and it also shows increased compactness in course of picoseconds time scale (in form of snapshots).
Conclusion
Drug repurposing has been proved as an effective method for predicting drug molecule bypassing the time consuming trial phases as because in this process the drugs were selected from FDA approved drug list. In this study, drug repurposing approach has been used to find out possible inhibitor of the enzyme Anhydro N acetylmuramic acid kinase and ergotamine was found to act as inhibitor for this protein. Ergotamine was previously used for migraine and now it has been proposed for the treatment of MDR Acinetobacter baumannii. In future, wet lab experiments can be performed for verifying the action of this repurposed drug on Acinetobacter baumannii.
Acknowledgment
The authors would like to acknowledge the Head, Department of Microbiology, The University of Burdwan for providing the infrastructure (computer lab of the department) for this work.
Conflict of Interest
he authors declare no conflict of interest.
Funding Source
No financial assistance was provided for carrying out this study.
References
- https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed.
- Fournier PE, Richet H, Weinstein RA. The epidemiology and control of Acinetobacter baumannii in health care facilities. Clin Infect Dis. 2006;42(5):692-9.
CrossRef - Howard A, O’Donoghue M, Feeney A, Sleator RD. Acinetobacter baumannii: an emerging opportunistic pathogen. 2012;3(3):243-50.
CrossRef - Lolans K, Rice TW, Munoz-Price LS, Quinn JP. Multicity outbreak of carbapenem-resistant Acinetobacter baumannii isolates producing the carbapenemase OXA-40. Antimicrobial agents and chemotherapy. 2006;50(9):2941-5.
CrossRef - Fishbain J, Peleg AY. Treatment of Acinetobacter Clin Infect Dis. 2010;51(1):79-84.
CrossRef - Paterson DL. The epidemiological profile of infections with multidrug-resistant Pseudomonas aeruginosa and Acinetobacter Clin Infect Dis. 2006; 43(Supplement_2):S43-8.
CrossRef - Hsueh PR, Teng LJ, Chen CY, Chen WH, Ho SW, Luh KT. Pandrug-resistant Acinetobacter baumannii causing nosocomial infections in a university hospital, Taiwan. Emerg Infect Dis. 2002;8(8):827.
CrossRef - Peleg AY, Adams J, Paterson DL. Tigecycline efflux as a mechanism for non susceptibility in Acinetobacter baumannii. Antimicrobial agents and chemotherapy. 2007;51(6):2065-9.
CrossRef - Kyriakidis I, Vasileiou E, Pana ZD, Tragiannidis A. Acinetobacter baumannii antibiotic resistance mechanisms. Pathogens. 2021;10 (3):373.
CrossRef - Jarada TN, Rokne JG, Alhajj R. A review of computational drug repositioning: strategies, approaches, opportunities, challenges, and directions. J cheminformatics. 2020;12(1):1-23.
CrossRef - Chen IM, Chu K, Palaniappan K, Ratner A, Huang J, Huntemann M, Hajek P, Ritter SJ, Webb C, Wu D, Varghese NJ. The IMG/M data management and analysis system v. 7: content updates and new features. Nucleic Acids Res. 2023;51(D1):D723-32.
CrossRef - Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403-10.
CrossRef - Gasteiger, E et al. Protein identification and Analysis Tools on the ExPASy server. In: Walker, J.M. (eds) The Proteomics Protocols Handbooks. Humana Press. 2005; pp 571-607.
CrossRef - YuN WJ, Laird M, Melli G, Rey S, Lo R, Dao P, Sahinalp S, Ester M, Foster L, Brinkman FP. 3.0: improved protein sub cellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. J Bioinform. 2017;26:1608-5.
- Mooney C, Wang YH, Pollastri G. SCLpred: protein sub cellular localization prediction by N-to-1 neural networks. J Bioinform. 2011;27(20):2812-9.
CrossRef - Puigbò P, Bravo IG, Garcia-Vallve S. CAIcal: a combined set of tools to assess codon usage adaptation. Biol direct. 2008;3(1):1-8.
CrossRef - Madeira F, Pearce M, Tivey AR, Basutkar P, Lee J, Edbali O, Madhusoodanan N, Kolesnikov A, Lopez R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022;50(W1):W276-9.
CrossRef - Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, Hachilif R, Gable AL, Fang T, Doncheva NT, Pyysalo S, Bork P. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51(D1):D638-46.
CrossRef - Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TA, Rempfer C, Bordoli L, Lepore R. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296-303.
CrossRef - Zhou X, Zheng W, Li Y, Pearce R, Zhang C, Bell EW, Zhang G, Zhang Y. I-TASSER-MTD: a deep-learning-based platform for multi-domain protein structure and function prediction. Nat Protoc. 2022;17(10):2326-53.
CrossRef - DeLano WL. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002;40(1):82-92.
- Colovos C, Yeates TO. Verification of protein structures: patterns of non bonded atomic interactions. Protein Sci. 1993;2(9):1511-9.
CrossRef - Eisenberg D, Bowie JU, Lüthy R, Choe S. Three-dimensional profiles for analysing protein sequence–structure relationships. Faraday Discuss. 1992;93:25-34.
CrossRef - Pontius J, Richelle J, Wodak SJ. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J Mol Biol. 1996;264(1):121-36.
CrossRef - Vriend G. WHAT IF: a molecular modelling and drug design program. J Mol graph. 1990;8(1):52-6.
CrossRef - Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereo chemical quality of protein structures. J Appl crystallogr. 1993;26(2):283-91.
CrossRef - Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic acids Res. 2018;46(W1):W363-7.
CrossRef - Jakubec D, Skoda P, Krivak R, Novotny M, Hoksza D. PrankWeb 3: accelerated ligand-binding site predictions for experimental and modelled protein structures. Nucleic Acids Res. 2022;50(W1):W593-7.
CrossRef - Liu Y, Yang X, Gan J, Chen S, Xiao ZX, Cao Y. CB-Dock2: Improved protein–ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022;50(W1):W159-64.
CrossRef - Sterling T, Irwin JJ. ZINC 15–ligand discovery for everyone. J chem inf model. 2015;55(11):2324-37.
CrossRef - Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. In: Hempel, J., Williams, C., Hong, C. (eds) Chemical Biology. Methods in Molecular Biology. Humana Press, 2015; pp 243-50.
CrossRef - Hospital A, Andrio P, Fenollosa C, Cicin-Sain D, Orozco M, Gelpí JL. MDWeb and MDMoby: an integrated web-based platform for molecular dynamics simulations. J Bioinform. 2012;28(9):1278-9.
CrossRef - Ikai A. Thermostability and aliphatic index of globular proteins. J Biochem. 1980;88(6):1895-8.
- Arun PP, Bakku RK, Subhashini M, Singh P, Prabhu NP, Suzuki I, Prakash JS. CyanoPhyChe: a database for physic-chemical properties, structure and biochemical pathway information of cyanobacterial proteins. PLoS One. 2012;7(11):e49425.
CrossRef - Mihelič M, Vlahoviček-Kahlina K, Renko M, Mesnage S, Doberšek A, Taler-Verčič A, Jakas A, Turk D. The mechanism behind the selection of two different cleavage sites in NAG-NAM polymers. IUCrJ. 2017;4(2):185-98.
CrossRef - Bacik JP, Whitworth GE, Stubbs KA, Yadav AK, Martin DR, Bailey-Elkin BA, Vocadlo DJ, Mark BL. Molecular basis of 1, 6-anhydro bond cleavage and phosphoryl transfer by Pseudomonas aeruginosa 1, 6-anhydro-N-acetylmuramic acid kinase. J Biol Chem. 2011;286(14):12283-91.
CrossRef - LENNOX WG. The use of ergotamine tartrate in migraine. N Engl J Med.. 1934;210(20):1061-5.
CrossRef - Kwarteng A, Asiedu E, Sylverken A, Larbi A, Mubarik Y, Apprey C. In silico drug repurposing for filarial infection predicts nilotinib and paritaprevir as potential inhibitors of the Wolbachia 5′-aminolevulinic acid synthase. Sci Rep. 2021;11(1):8455.
CrossRef - Lobanov MY, Bogatyreva NS, Galzitskaya OV. Radius of gyration as an indicator of protein structure compactness. Mol Biol. 2008;42:623-8.
CrossRef
Accepted on: 22-11-2023
Second Review by: Dr. Joel Omage
Final Approval by: Dr. Imran Ali








![]()
![]()

{kind=link}