
In Silico Design and ADMET Profiling of Thiadiazole Derivatives Targeting NUDT5 (5NQR) for Breast Cancer Therapy
 , Rohit Jaysing Bhor1* , Amol Mohan Shirode2, Pravin Bapurao Jadhav3 , Rutuja Rajendra Lokhande1, Prasad Jagdish Mantode1
, Rohit Jaysing Bhor1* , Amol Mohan Shirode2, Pravin Bapurao Jadhav3 , Rutuja Rajendra Lokhande1, Prasad Jagdish Mantode1 1Department of Pharmaceutical Chemistry, Pravara Rural College of Pharmacy, Ahmednagar, India
2Department of Pharmaceutical Chemistry, K. B. H. S. S. Trust Institute of Pharmacy, Nashik, India
3Department of Pharmaceutical Chemistry, Sandip Institute of Pharmaceutical sciences, Nashik, Maharashtra, India.
Corresponding Author E-mail: rohit.bhor69@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3540
Download this article as:
![]()
The present investigation focuses on the in-silico evaluation of thiadiazole-based substituted derivatives as potential inhibitors of the 5NQR protein, NUDT5, a promising molecular target associated with breast cancer progression. NUDT5 is involved in nuclear ATP synthesis, chromatin remodelling, and hormone-regulated breast cancer growth, making it an attractive target for anticancer drug discovery. A library of designed compounds (VB1–VB12) was subjected to molecular docking studies to evaluate their binding affinity and interactions within the active site of the target protein. Docking analysis revealed favorable ligand–protein interactions, with binding energies ranging from −8.6 to −10.5 kcal/mol. Among the tested compounds, VB11, VB1, and VB10 exhibited the strongest binding affinities, supported by multiple hydrogen bonds, hydrophobic interactions, and electrostatic interactions with key active-site amino acid residues. Lead compound selection was based on an integrated evaluation of docking affinity, ADMET properties, drug-likeness, and pharmacokinetic suitability rather than docking score alone. Although VB11 and VB1 demonstrated superior docking scores, compounds VB8, VB2, and VB7 showed more favorable overall pharmacokinetic and drug-likeness characteristics, including lower rule violations, improved predicted gastrointestinal absorption, safer CYP inhibition profiles, and balanced physicochemical properties. Physicochemical and drug-likeness analyses indicated that most compounds complied with standard medicinal chemistry parameters, supporting their suitability for further drug development. Based on the combined findings from molecular docking, pharmacokinetic profiling, and drug-likeness evaluation, VB8, VB2, and VB7 were identified as the most promising lead candidates. In this study suggests that thiadiazole derivatives may serve as valuable scaffolds for the development of novel anticancer agents targeting NUDT5 and warrants further in-vitro and in-vivo investigations.
KEYWORDS:Abbreviations ADME: Absorption, Distribution, Metabolism, and Excretion ADMET: Absorption, Distribution, Metabolism, Excretion, and Toxicity BBB: Blood–Brain Barrier CYP: Cytochrome P450 GI: Gastrointestinal HBA: Hydrogen Bond Acceptor HBD: Hydrogen Bond Donor MR: Molar Refractivity NUDT5: Nudix Hydrolase 5 PDB: Protein Data Bank P-gp: P-glycoprotein TPSA: Topological Polar Surface Area
Introduction
Cancer remains a major global health concern characterized by uncontrolled cell proliferation, evasion of apoptosis, tissue invasion, and metastasis. Its development is driven by genetic mutations,1 epigenetic alterations,2 and dysregulation of cellular signaling pathways. Chronic inflammation is increasingly recognized as an important contributor to cancer initiation and progression. Among the molecular targets associated with cancer, NUDT5 (Nudix hydrolase 5) has gained attention due to its role in ADP-ribose metabolism,3 nuclear ATP synthesis, chromatin remodeling, and hormone-dependent transcriptional regulation in breast cancer. Overexpression of NUDT5 has been reported in several malignancies, including breast, colorectal,4 lung, and prostate cancers, highlighting its therapeutic significance. Advances in molecular biology have improved the understanding of breast cancer pathogenesis and facilitated the development of targeted therapies. In this context, the 5NQR protein structure of NUDT5 serves as an important target for anticancer drug discovery. Molecular docking is a widely used computational approach in structure-based drug design that predicts ligand binding orientation and estimates binding affinity within the active site of target proteins. The method employs search algorithms and scoring functions to evaluate ligand–protein interactions,5 including hydrogen bonding,6 hydrophobic interactions,7 van der Waals forces,8 and electrostatic contacts. Lower docking energy values generally indicate stronger and more stable binding interactions. Molecular docking has become an essential tool in cancer research because it enables rapid screening of compounds,9 reduces experimental cost and time, and provides insight into molecular interaction mechanisms. Commonly used software platforms include AutoDock,10 PyRx,11 and BIOVIA Discovery Studio.12 Despite limitations such as restricted protein flexibility and dependence on scoring accuracy,13 docking studies significantly support rational drug design and lead optimization for the development of safer and more effective anticancer agents targeting NUDT5. Despite these promising computational findings,14 several limitations should be acknowledged. ADMET predictions are based on statistical and machine-learning models and therefore remain approximations rather than experimentally validated pharmacokinetic outcomes. Similarly, docking scores and interaction energies may vary depending on scoring algorithms and receptor preparation methods. Possible off-target interactions,15 toxicity risks,16 and long-term biological effects were not evaluated in the present study. Furthermore, the absence of molecular dynamics simulations limits understanding of the stability and flexibility of ligand–protein complexes over time under physiological conditions.
Materials and Methods
Materials
The link between the target receptor, the anticipated lead chemical molecule,17 and its protein receptors is often investigated using docking in modern drug research. The study was conducted electronically using computational techniques. ADMET Prediction in Silico to forecast their pharmacokinetic profiles,18 the best medications selected from molecular docking studies were evaluated for ADME characteristics. The sketch tool in Discovery Studio is used to create the molecules in two and three dimensions. The compound was created in Marvin Sketch,19 optimized in three dimensions, and saved in SDF format.
Methods
Protein Preparation
The 5NQR | pdb_00005nqr protein corresponds to the crystal structure of NUDT5 (Nudix hydrolase 5), an enzyme that has gained significant attention for its role in cancer metabolism and nuclear signaling. NUDT5 belongs to the Nudix hydrolase family,20 which is involved in the hydrolysis of nucleoside diphosphate derivatives, thereby regulating intracellular nucleotide pools and maintaining cellular homeostasis. Recent studies have identified NUDT5 as a critical regulator of nuclear ATP synthesis,21 particularly in hormone-responsive cancers such as breast cancer. Unlike conventional ATP production pathways that occur in mitochondria, NUDT5 facilitates the generation of ATP within the nucleus through the hydrolysis of ADP-ribose. This localized ATP production is essential for energy-dependent chromatin remodeling processes,22 which enable the transcriptional activation of genes involved in cell proliferation and survival. The three-dimensional crystal structure of inhibitor (5NQR | pdb_00005nqr) was provided by the RCSB Protein Data Bank with mention in Figure 1.
 |
Figure 1: 5NQR | pdb_00005nqr protein corresponds to the crystal structure of NUDT5 (Nudix hydrolase 5), an enzyme that has gained significant attention for its role in cancer metabolism.
|
Preparation of ligand
1,3,4-thiadiazole and its substituted scaffolds were used as ligands mention in Figure 2 for the docking study. For chemical structures,23 Chem Draw was used to sketch, which were then converted into three-dimensional structures. Ligands were prepared by using Schrodinger’s Lig Prep module. The ligands’ overall energy-minimized Potential of stereoisomers were kept,24 and the refined ligands were stored in Maestro format.25 The derivatives of 1,3,4-thiadiazole and its substituted scaffolds were used as ligands mention in Figure 2 for the docking study.
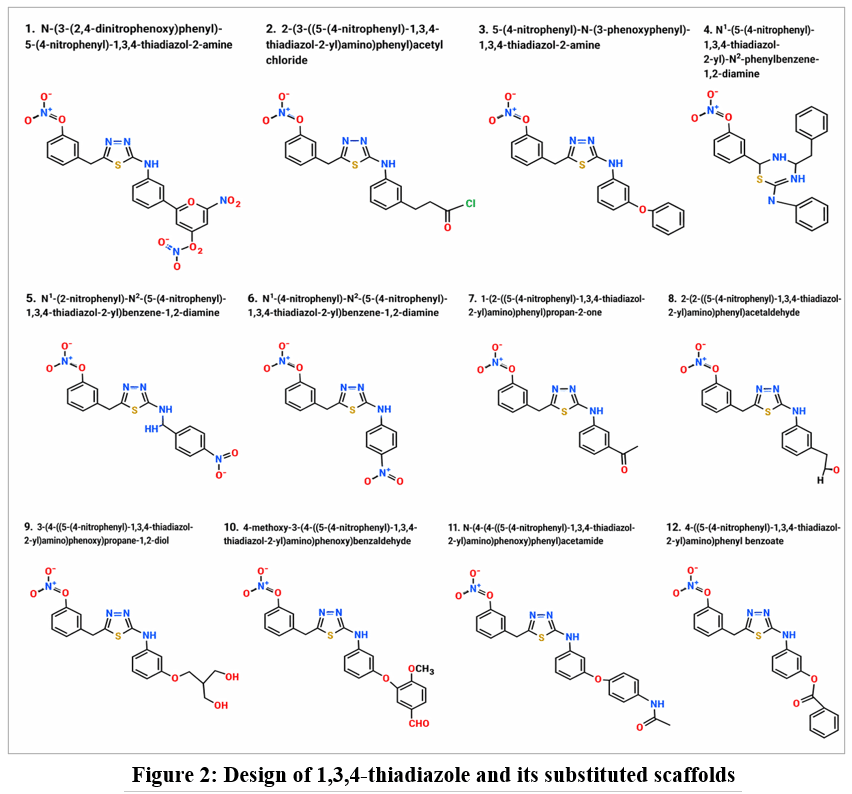 |
Figure 2: Design of 1,3,4-thiadiazole and its substituted scaffolds
|
Results
Physicochemical Property Analysis of Designed Molecules (VB1–VB12)
The physicochemical properties of compounds VB1–VB12 were evaluated to assess their drug-likeness and suitability for oral administration, including parameters such as rotatable bonds, hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), molar refractivity (MR), topological polar surface area (TPSA), and lipophilicity. The detailed results are presented in Table 1 and Figure 3. Most compounds exhibited moderate molecular flexibility, with VB1, VB9, VB10, and VB11 showing higher flexibility due to increased rotatable bonds. HBA values ranged from 4–9 and HBD values from 1–3, indicating varying hydrogen-bonding capacities that may influence target binding and membrane permeability. The MR values (93.68–127.87) suggested acceptable molecular volume and polarizability, while TPSA values ranged from 121.1–212.74 Ų. Compounds VB3 and VB4 showed TPSA values favorable for oral bioavailability, whereas VB1 exhibited very high polarity, which may reduce membrane permeability. Lipophilicity analysis using iLOGP, XLOGP3, WLOGP, and MLOGP indicated moderate to high lipophilicity among the compounds, with VB5 showing the highest value. Overall, compounds VB7, VB8, and VB2 demonstrated balanced physicochemical characteristics, including moderate TPSA, acceptable lipophilicity, and suitable hydrogen-bonding properties, suggesting favorable drug-like behavior compared to the more polar or highly lipophilic compounds VB1, VB5, VB6, and VB9.
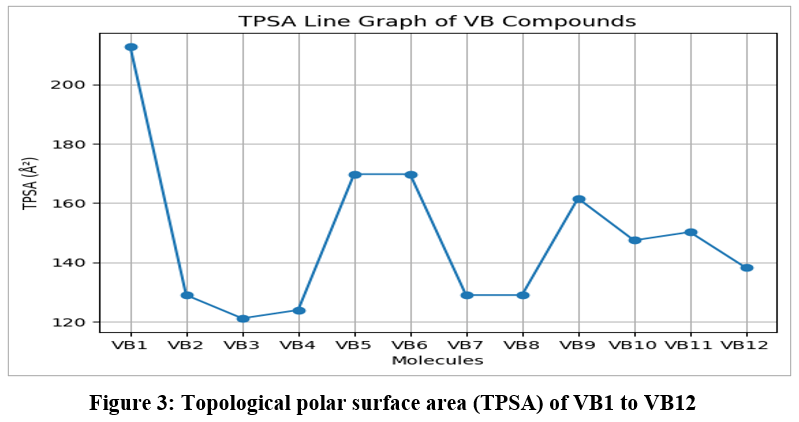 |
Figure 3: Topological polar surface area (TPSA) of VB1 to VB12
|
Table 1: Comparative table containing #Rotatable bonds; #H-bond acceptors; #H-bond donors; MR; TPSA; iLOGP; XLOGP3; WLOGP and MLOGP for VB1 to VB12
| Molecule | #Rotatable bonds | #H-bond acceptors | #H-bond donors | MR | TPSA | iLOGP | XLOGP3 | WLOGP | MLOGP |
| VB1 | 8 | 9 | 1 | 127.87 | 212.74 | 2.2 | 4.92 | 5.47 | 2.1 |
| VB2 | 6 | 5 | 1 | 98.48 | 128.94 | 1.91 | 4.18 | 4.16 | 2.64 |
| VB3 | 6 | 5 | 1 | 110.23 | 121.1 | 3.03 | 5.26 | 5.65 | 3.73 |
| VB4 | 6 | 4 | 2 | 113.25 | 123.9 | 2.73 | 5.27 | 5.6 | 3.73 |
| VB5 | 7 | 6 | 2 | 122.08 | 169.72 | 2.98 | 5.65 | 5.51 | 2.88 |
| VB6 | 7 | 6 | 2 | 122.08 | 169.72 | 2.42 | 5.1 | 5.51 | 2.88 |
| VB7 | 6 | 5 | 1 | 98.49 | 128.94 | 1.9 | 3.35 | 3.99 | 2.64 |
| VB8 | 6 | 5 | 1 | 93.68 | 128.94 | 1.49 | 3.13 | 3.6 | 2.4 |
| VB9 | 8 | 7 | 3 | 102.14 | 161.56 | 2.11 | 2.4 | 2.59 | 0.6 |
| VB10 | 8 | 7 | 1 | 122.11 | 147.4 | 2.67 | 4.69 | 5.47 | 1.73 |
| VB11 | 8 | 6 | 2 | 124.54 | 150.2 | 2.72 | 4.44 | 5.42 | 2.13 |
| VB12 | 7 | 6 | 1 | 115.11 | 138.17 | 2.67 | 5.14 | 5.08 | 2.83 |
ADME and Pharmacokinetic Profile of Designed Molecules (VB1–VB12)
The physicochemical properties of compounds VB1–VB12 were evaluated to determine their drug-likeness and oral bioavailability using parameters such as rotatable bonds, hydrogen bond acceptors/donors (HBA/HBD), molar refractivity (MR), topological polar surface area (TPSA), and lipophilicity. Most compounds showed moderate molecular flexibility, while VB1, VB9, VB10, and VB11 exhibited comparatively higher flexibility. HBA values ranged from 4–9 and HBD values from 1–3, indicating moderate hydrogen-bonding potential. MR values (93.68–127.87) suggested acceptable molecular volume and polarizability. TPSA values ranged from 121.1–212.74 Ų, with VB3 and VB4 showing favorable oral bioavailability, whereas VB1 displayed high polarity that may reduce membrane permeability. Lipophilicity analysis revealed moderate to high logP values, with VB5 exhibiting the highest lipophilicity. Overall, VB7, VB8, and VB2 demonstrated balanced physicochemical properties and favorable drug-like characteristics compared to the more polar or highly lipophilic compounds VB1, VB5, VB6, and VB9.
Table 2: Comparative table containing GI absorption; BBB permeant; Pgp substrate; CYP1A2 inhibitor; CYP2C19 inhibitor; CYP2C9 inhibitor; CYP2D6 inhibitor; CYP3A4 inhibitor for VB1–VB12
| Molecule | GI absorption | BBB permeant | Pgp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor |
| VB1 | Low | No | No | Yes | Yes | Yes | No | Yes |
| VB2 | Low | No | No | Yes | Yes | Yes | No | No |
| VB3 | Low | No | No | Yes | Yes | Yes | No | No |
| VB4 | Low | No | No | Yes | Yes | Yes | Yes | Yes |
| VB5 | Low | No | No | Yes | Yes | Yes | No | Yes |
| VB6 | Low | No | No | Yes | Yes | Yes | No | Yes |
| VB7 | Low | No | No | Yes | Yes | Yes | No | No |
| VB8 | High | No | No | Yes | Yes | Yes | No | No |
| VB9 | Low | No | Yes | No | No | Yes | Yes | Yes |
| VB10 | Low | No | No | No | Yes | Yes | No | Yes |
| VB11 | Low | No | No | No | Yes | Yes | No | Yes |
| VB12 | Low | No | No | No | Yes | Yes | No | Yes |
Drug-Likeness Evaluation of Designed Molecules (VB1–VB12)
The drug-likeness properties of compounds VB1–VB12 were evaluated using Lipinski, Ghose, Veber, Egan, and Muegge filters along with bioavailability score prediction, as presented in Table 3. Most compounds showed good compliance with Lipinski’s rule of five, with only VB1 exhibiting a single violation. Minor deviations in the Ghose filter were observed for VB1, VB3, and VB4, while the remaining compounds satisfied all criteria.According to Veber’s and Egan’s rules, compounds VB1, VB5, VB6, VB9, VB10, and VB11 showed violations related to molecular flexibility, polarity, or permeability, which may affect oral absorption. The Muegge filter identified VB5 and VB6 as having the highest number of violations, suggesting weaker drug-like characteristics. In contrast, VB2, VB7, VB8, and VB10 fully complied with the Muegge criteria and demonstrated favorable drug-like profiles. Overall, VB2, VB7, and VB8 emerged as the most promising candidates due to their minimal rule violations and acceptable bioavailability scores, whereas VB5 and VB6 may require structural optimization to improve their pharmacokinetic properties.
Table 3: Comparative table containing Lipinski #violations; Ghose #violations; Veber #violations; Egan #violations; Muegge #violations and Bioavailability Score for VB1–VB12
| Molecule | Lipinski #violations | Ghose #violations | Veber #violations | Egan #violations | Muegge #violations | Bioavailability Score |
| VB1 | 1 | 1 | 1 | 1 | 1 | 0.55 |
| VB2 | 0 | 0 | 0 | 0 | 0 | 0.55 |
| VB3 | 0 | 1 | 0 | 0 | 1 | 0.55 |
| VB4 | 0 | 1 | 0 | 0 | 1 | 0.55 |
| VB5 | 0 | 0 | 1 | 1 | 2 | 0.55 |
| VB6 | 0 | 0 | 1 | 1 | 2 | 0.55 |
| VB7 | 0 | 0 | 0 | 0 | 0 | 0.55 |
| VB8 | 0 | 0 | 0 | 0 | 0 | 0.55 |
| VB9 | 0 | 0 | 1 | 1 | 1 | 0.55 |
| VB10 | 0 | 0 | 1 | 1 | 0 | 0.55 |
| VB11 | 0 | 0 | 1 | 1 | 1 | 0.55 |
| VB12 | 0 | 0 | 0 | 1 | 1 | 0.55 |
Molecular Docking Interaction Analysis of Designed Compounds (VB Series)
The molecular docking analysis of the designed compounds (VB1–VB12), as presented in Figure 4; demonstrated notable interactions with critical active-site residues of the target protein, indicating strong binding affinity and stable ligand–protein complex formation. The calculated docking scores ranged from −8.6 to −10.5 kcal/mol, reflecting generally interaction energies across the compound series. Among the evaluated molecules, VB11 exhibited the highest binding affinity with a docking score of −10.5 kcal/mol, followed by VB1 (−10.2 kcal/mol) and VB10 (−9.8 kcal/mol), suggesting the formation of highly stable complexes within the active site. In contrast, VB7 showed the lowest binding affinity (−8.6 kcal/mol), although its score still falls within a range indicative of potential biological activity. Hydrogen bonding interactions were found to play a key role in stabilizing the ligand–protein complexes. Several active-site residues, including ARG51, PHE167, GLN15, GLY135, and GLU166, were consistently involved in conventional hydrogen bond formation across multiple ligands. The observed bond lengths, ranging from approximately 1.07 Å to 2.83 Å, indicate the presence of strong and energetically hydrogen bonds. Notably, compounds VB1 and VB3 established multiple hydrogen bonding interactions with residues such as ARG51, PHE167, and GLN15, which likely contributes to enhanced binding specificity and stability. Furthermore, VB8 displayed the highest number of hydrogen bond interactions, including both conventional and carbon–hydrogen bonds, supporting its stable binding conformation within the active site.VB2 demonstrated very short hydrogen bond distances (~1.07 Å), suggesting particularly strong interactions. Electrostatic interactions, including π-cation and π-anion interactions, were observed with residues such as ARG51, ARG84, and GLU166, which significantly contribute to binding strength. These interactions were particularly prominent in VB1, VB2, VB3, VB4, VB9, VB10, and VB11. Additionally, π–π stacking and π–alkyl interactions were frequently observed with aromatic residues such as TRP28, TRP46, TYR36, and PHE167. These hydrophobic interactions enhance ligand stabilization within the binding pocket: VB7 and VB8 showed extensive π–π stacking interactions with tryptophan residues. VB9, VB10, and VB11 demonstrated multiple π-alkyl and π-sigma interactions with hydrophobic residues such as LEU98, ILE141, and MET132. Hydrophobic interactions were widely distributed across all compounds, involving residues such as LEU98, ALA96, ILE141, and MET132. These interactions play a critical role in maintaining ligand orientation and improving binding affinity within the hydrophobic regions of the protein. Overall, compounds VB11, VB1, and VB10 demonstrated superior docking performance due to a combination of strong hydrogen bonding, electrostatic interactions, and extensive hydrophobic contacts. Although VB8 exhibited slightly lower docking scores, its high number of hydrogen bonds and balanced interaction profile suggest good binding stability. Conversely, VB7 and VB9 showed relatively lower binding affinities, which may be attributed to weaker or fewer stabilizing interactions despite the presence of multiple hydrophobic contacts.
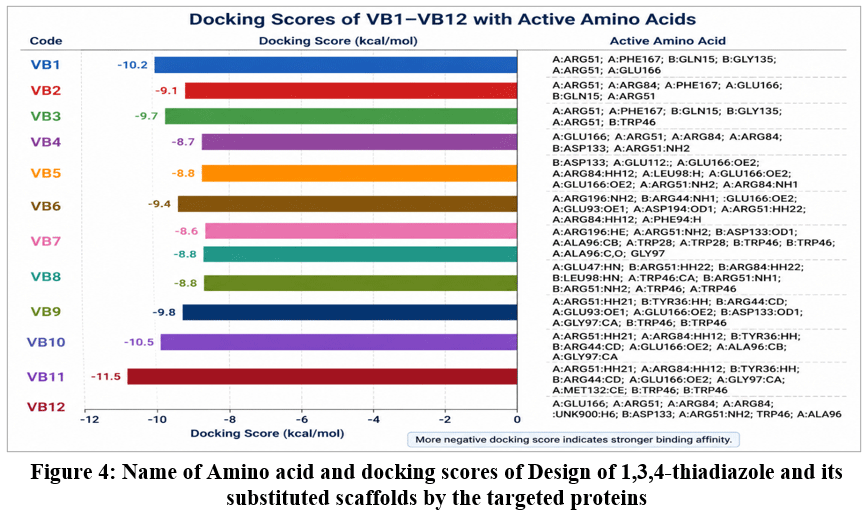 |
Figure 4: Name of Amino acid and docking scores of Design of 1,3,4-thiadiazole and its substituted scaffolds by the targeted proteins
|
Although compounds VB12, VB11, and VB1 exhibited stronger docking affinities toward the NUDT5 active site, lead prioritization was performed using an integrated evaluation approach that included docking interactions, ADMET profile, physicochemical characteristics, drug-likeness parameters, gastrointestinal absorption, CYP450 inhibition profile, and overall pharmacokinetic suitability. Among the evaluated compounds, VB8, VB2, and VB7 demonstrated comparatively balanced properties with favorable drug-likeness, minimal rule violations, safer predicted metabolic profiles, and improved pharmacokinetic characteristics. In particular, VB8 showed high predicted gastrointestinal absorption and lower CYP3A4 inhibitory potential, making it pharmacokinetically more favorable despite a comparatively lower docking score.
Discussion
The present investigation demonstrates the usefulness of integrating molecular docking, physicochemical evaluation, ADMET prediction, drug-likeness screening, and energy minimization studies for the identification of potential NUDT5-targeted compounds. NUDT5 is an important therapeutic target in breast cancer due to its role in nuclear ATP synthesis, chromatin remodeling, and hormone-dependent cell proliferation. The designed 1,3,4-thiadiazole derivatives showed favorable interaction profiles with the active site of NUDT5, with VB11, VB1, and VB10 exhibiting the strongest docking affinities. Key amino acid residues such as ARG51, GLU166, PHE167, and TRP46 contributed significantly to ligand stabilization through hydrogen bonding, electrostatic, and hydrophobic interactions. However, docking affinity alone cannot reliably predict biological activity. Although VB11, VB1, and VB10 demonstrated superior docking scores, compounds VB8, VB2, and VB7 were prioritized as lead candidates based on their balanced pharmacokinetic, physicochemical, and drug-likeness properties. In particular, VB8 exhibited favorable gastrointestinal absorption, balanced lipophilicity, fewer drug-likeness violations, and safer CYP450 interaction profiles, suggesting improved pharmacokinetic suitability. Physicochemical analysis indicated that most compounds possessed acceptable molecular descriptors, while highly polar compounds such as VB1, VB5, VB6, and VB9 may show reduced membrane permeability. ADMET studies further revealed generally low blood–brain barrier permeability and comparatively safer metabolic profiles for VB2, VB7, and VB8. Energy minimization studies supported the structural stability of VB2, VB3, and VB7, whereas VB4, VB5, and VB6 may require additional structural optimization. Molecular docking analysis revealed appreciable binding affinities across the compound series, with VB11, VB1, and VB10 exhibiting the strongest docking scores. Detailed interaction analysis showed that amino acid residues such as ARG51, GLU166, PHE167, and TRP46 contributed significantly to ligand stabilization through hydrogen bonding, electrostatic interactions, and hydrophobic contacts. These interactions suggest that the 1,3,4-thiadiazole scaffold may provide a structurally suitable framework for interaction with the catalytic region of NUDT5. However, strong docking affinity alone cannot be considered definitive evidence of anticancer activity or inhibitory efficacy. Molecular docking provides only a predictive estimate of ligand–protein interaction under simplified computational conditions and does not fully account for protein flexibility, solvent effects, dynamic conformational changes, or complex biological environments. Therefore, compounds with strong docking scores may not necessarily demonstrate superior biological activity under experimental conditions. An important observation in the present study was the difference between docking-based ranking and pharmacokinetic prioritization. Overall, the study identifies VB8, VB2, and VB7 as promising preliminary lead compounds for NUDT5-targeted anticancer drug development. Nevertheless, these findings are predictive and require further validation through molecular dynamics simulations, in-vitro studies, in-vivo investigations, and experimental pharmacological evaluation.
Conclusion
In conclusion, the present in-silico study indicates that the designed 1,3,4-thiadiazole derivatives possess promising potential as NUDT5-targeted anticancer agents. Molecular docking studies demonstrated favorable ligand–protein interactions, with compounds such as VB11 and VB12 exhibiting strong binding affinities toward the active site of NUDT5. However, lead prioritization was based on integrated evaluation of docking results, physicochemical parameters, ADMET properties, drug-likeness, and energy minimization studies rather than docking score alone. Among the evaluated compounds, VB8, VB2, and VB7 emerged as the most promising lead candidates due to their balanced physicochemical and pharmacokinetic profiles. These compounds showed acceptable lipophilicity, moderate TPSA values, minimal drug-likeness rule violations, and favorable ADMET characteristics. In particular, VB8 demonstrated comparatively higher predicted gastrointestinal (GI) absorption, balanced polarity, and safer CYP450 inhibition profiles, suggesting improved oral bioavailability and pharmacokinetic suitability. Additionally, most compounds were predicted to possess low blood–brain barrier permeability and non-substrate behavior toward P-glycoprotein, which may reduce central nervous system exposure and efflux-related complications. Overall, the findings suggest that the 1,3,4-thiadiazole scaffold may serve as a useful framework for the development of novel NUDT5 inhibitors. Nevertheless, the present results are predictive in nature and require further validation through molecular dynamics simulations, in-vitro assays, in-vivo studies, and experimental pharmacological investigations to confirm their biological activity, safety, and therapeutic efficacy.
Acknowledgement
The authors are thankful to Pravara Rural College of Pharmacy, Pravaranagar.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable.
Author Contributions:
- Vaishnavi Babasaheb Bagale: Contribute in In-silico ADMET Data
- Rohit Jaysing Bhor: The active amino residues, bond length, bond category, bond type, ligand energies
- Amol Mohan Shirode: Drawing of drug derivatives
- Pravin Bapurao Jadhav: SWISS ADMET Software Data
- Rutuja Rajendra Lokhande: Docking images of 2D and 3D by Discovery Studio 2021
- Prasad Jagdish Mantode: Research drafting
References
- Page BDG, Valerie NCK, Wright RHG, et al. Targeted NUDT5 inhibitors block hormone signaling in breast cancer cells. Nature Communications. 2018;9(1):250. doi:10.1038/s41467-017-02293-7.
CrossRef - Sultana R, Islam M, Haque MA, et al. Molecular docking based virtual screening of the breast cancer target NUDT5. Bioinformation. 2019;15(11):784-789. doi:10.6026/97320630015784.
CrossRef - Ruswanto R, Nofianti T, Mardianingrum R, Kesuma D, Siswandono. Design, molecular docking, and molecular dynamics of thiourea-iron (III) metal complexes as NUDT5 inhibitors for breast cancer treatment. Heliyon. 2022;8(9):e10694. doi:10.1016/j.heliyon.2022.e10694.
CrossRef - Dhanasekaran S, Pradeepkumar PI, Rajasekaran R, et al. Targeting Nudix Hydrolase 5 with Bioactive Flavonoids: Molecular Dynamics and Docking Studies for Breast Cancer Therapy. Cell Biochemistry and Biophysics. 2025;83(2):1973-1991. doi:10.1007/s12013-024-01609-x.
CrossRef - Ramasamy S, Jeyaram K, Narayanan A, et al. Repurposing fluvoxamine as an inhibitor for NUDT5 in breast cancer cell: an in silico and in vitro study. Drug Research. 2025;13(1):5. doi:10.1007/s40203-024-00293-2.
CrossRef - Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry. 2010;31(2):455-461. doi:10.1002/jcc.21334.
CrossRef - Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. In: Chemical Biology. Humana Press; 2015:243-250. doi:10.1007/978-1-4939-2269-7_19.
CrossRef - Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785-2791. doi:10.1002/jcc.21256.
CrossRef - Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 2001;46(1-3):3-26. doi:10.1016/S0169-409X(00)00129-0.
CrossRef - Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence oral bioavailability of drug candidates. Journal of Medicinal Chemistry. 2002;45(12):2615-2623. doi:10.1021/jm020017n.
CrossRef - Egan WJ, Merz KM Jr, Baldwin JJ. Prediction of drug absorption using multivariate statistics. Journal of Medicinal Chemistry. 2000;43(21):3867-3877. doi:10.1021/jm000292e.
CrossRef - Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. Journal of Combinatorial Chemistry. 1999;1(1):55-68. doi:10.1021/cc9800071.
CrossRef - Muegge I, Heald SL, Brittelli D. Simple selection criteria for drug-like chemical matter. Journal of Medicinal Chemistry. 2001;44(12):1841-1846. doi:10.1021/jm015507e.
CrossRef - Ferreira LG, dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20(7):13384-13421. doi:10.3390/molecules200713384.
CrossRef - Lionta E, Spyrou G, Vassilatis DK, Cournia Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Current Topics in Medicinal Chemistry. 2014;14(16):1923-1938. doi:10.2174/1568026614666140929124445.
CrossRef - Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Reviews Drug Discovery. 2004;3(11):935-949. doi:10.1038/nrd1549.
CrossRef - Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Current Computer-Aided Drug Design. 2011;7(2):146-157. doi:10.2174/157340911795677602.
CrossRef - Pagadala NS, Syed K, Tuszynski J. Software for molecular docking: a review. Biophysical Reviews. 2017;9(2):91-102. doi:10.1007/s12551-016-0247-1.
CrossRef - Pinheiro PSM, Justino GC. Structural analysis in silico of molecular properties, drug-likeness, and toxicity of curcumin analogues. Journal of Molecular Modeling. 2012;18(8):3699-3708. doi:10.1007/s00894-012-1389-7.
- DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. Journal of Health Economics. 2016;47:20-33. doi:10.1016/j.jhealeco.2016.01.012.
CrossRef - Bernstein FC, Koetzle TF, Williams GJ, et al. The Protein Data Bank: a computer-based archival file for macromolecular structures. Journal of Molecular Biology. 1977;112(3):535-542. doi:10.1016/S0022-2836(77)80200-3.
CrossRef - Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Research. 2000;28(1):235-242. doi:10.1093/nar/28.1.235.
CrossRef - Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Research. 2018;46(D1):D1074-D1082. doi:10.1093/nar/gkx1037.
CrossRef - Kim S, Chen J, Cheng T, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49(D1):D1388-D1395. doi:10.1093/nar/gkaa971.
CrossRef - Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296-W303. doi:10.1093/nar/gky427.
CrossRef
Accepted on: 21-05-2026
Second Review by: Dr. Nafees Ahamad
Final Approval by: Dr. Eugene A. Silow








![]()
![]()
