
IFN-I Signaling in SLE: Emerging Targets Beyond Conventional Therapy
 , Kishor Otari and Ajay Kale
, Kishor Otari and Ajay Kale Department of Pharmacology, Navsahyadri Institute of Pharmacy, Pune, India
Corresponding author E-mail: satavpriya3@gmail.com
Download this article as:
ABSTRACT:Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by immune dysregulation, autoantibody production, and multisystem inflammation. A hallmark feature of SLE is persistent activation of the type I interferon (IFN-I) pathway, commonly known as the interferon signature. Plasmacytoid dendritic cells are the major source of IFN-α and contribute to sustained immune activation through nucleic acid–containing immune complexes. IFN-I signaling enhances dendritic cell maturation, promotes T helper 1 polarization, and supports survival and activation of autoreactive B cells through BAFF and APRIL pathways. Therapeutic approaches targeting BAFF and IFNAR1 have demonstrated reductions in disease activity and flare frequency, particularly in patients with high interferon activity. However, infection risk remains an important limitation of these therapies. This review summarizes current evidence regarding IFN-I–mediated immune dysregulation in SLE and highlights the comparative therapeutic significance of BAFF and IFNAR1 pathways in the development of precision-based treatment strategies. Plasmacytoid dendritic cells (pDCs) are the main source of IFN-α in SLE. They become aberrantly activated by immune complexes containing nucleic acids derived from apoptotic debris. This activation sustains IFN-I signaling and drives a cascade of immune responses. These include dendritic cell maturation, T helper 1 (Th1) polarization, and the survival and expansion of autoreactive B cells. The IFN-I pathway also enhances B-cell activation through BLyS/BAFF and APRIL signaling, promoting increased autoantibody production. Together, these processes establish a self-amplifying autoimmune loop that contributes to disease progression. Therapeutic strategies targeting these pathways have shown encouraging clinical outcomes. Inhibition of BAFF and blockade of the interferon receptor subunit IFNAR1 have been associated with reductions in disease activity and flare frequency, along with improved control of organ involvement. However, these benefits must be balanced against an increased risk of infections, reflecting the immunomodulatory effects of these therapies. This review synthesizes current evidence on the role of type I interferons in SLE pathogenesis, with particular emphasis on the distinct yet complementary roles of BAFF and IFNAR1 signaling pathways. It also evaluates the efficacy and safety profiles of emerging biologic therapies. A clearer understanding of how innate and adaptive immune pathways are differentially dysregulated in SLE provides a strong foundation for the development of more precise, targeted treatment strategies.
KEYWORDS:Autoimmunity Targeted biologic therapy; Immunity; Plasmacytoid dendritic cells (pDCs); Systemic lupus erythematosus (SLE); Type I interferon (IFN-I)
Introduction
Systemic lupus erythematosus (SLE) is a chronic, multifaceted autoimmune disorder characterized by multisystem involvement and a highly variable clinical presentation. The disease results from a breakdown of immune tolerance, leading to the production of pathogenic autoantibodies, formation of immune complexes, and subsequent inflammation that damages multiple tissues. Organs commonly affected include the kidneys, skin, joints, cardiovascular system, and central nervous system, reflecting the systemic nature of the condition.1,4,9,16,17 Despite significant advances in research, the precise etiology of SLE remains incompletely understood. Current evidence suggests that disease onset and progression are influenced by a complex interaction of genetic susceptibility, epigenetic changes, hormonal factors, and environmental triggers.1,4,10
A central feature of SLE pathogenesis is the dysregulation of both innate and adaptive immune responses. Aberrant activation of the innate immune system promotes persistent inflammatory signaling and contributes to the activation of autoreactive lymphocytes. Among the various molecular pathways implicated, the type I interferon (IFN-I) system has emerged as a key driver of disease development. Patients with SLE frequently exhibit increased expression of interferon-stimulated genes, referred to as the “interferon signature,” which correlates closely with disease activity and severity. Sustained IFN-I signalling further amplifies immune dysregulation by promoting dendritic cell maturation, enhancing antigen presentation, and facilitating the expansion of autoreactive T and B lymphocytes.7,8,10
Plasmacytoid dendritic cells (pDCs) are recognized as the main source of type I interferons, particularly interferon-α, in SLE. These cells are activated by endogenous nucleic acids released from apoptotic or necrotic cells. The nucleic acids form complexes with autoantibodies and are detected by intracellular toll-like receptors, initiating a self-perpetuating cycle of interferon production, immune activation, and tissue injury.11,12,14 In addition, genetic variations affecting nucleic acid sensing pathways and interferon signaling have been strongly associated with increased susceptibility to SLE, further highlighting the importance of this pathway.10,13
The effects of IFN-I extend beyond innate immunity and significantly influence adaptive immune responses. It promotes B-cell activation, differentiation, and antibody production while impairing regulatory mechanisms that normally maintain immune tolerance. Consequently, a wide range of autoantibodies—including anti-dsDNA, anti-Ro/La, and anti-RNP—are produced, often years before the onset of clinical symptoms.21,22,24 These autoantibodies contribute to immune complex deposition in target organs, leading to complications such as lupus nephritis and cutaneous manifestations.17,23
Clinically, SLE shows marked heterogeneity, with disease manifestations ranging from mild mucocutaneous involvement to severe, life-threatening organ damage. This variability reflects differences in underlying immunological mechanisms among patients, particularly the extent of interferon pathway activation.4,9,18 Recognition of the IFN-I pathway as a critical contributor to disease pathogenesis has led to the development of targeted therapies, including interferon receptor antagonists and biologics such as belimumab, which have demonstrated promising outcomes in clinical studies.2,3,5,18,19
In summary, SLE is a multifactorial autoimmune disease driven by complex immune dysregulation, with the type I interferon pathway playing a central role in disease initiation and progression. A deeper understanding of these mechanisms provides valuable insight into disease heterogeneity and supports the development of more precise and targeted therapeutic approaches.15,20,25
Type I interferon pathway
Components of the type I interferon pathway include IFN-inducing stimuli, IFN-related genes and proteins, producing cells, and responding target cells. Humans possess the system comprises 15 functional IFN-I genes, including 13 IFN-α isoforms, subtypes, IFN-β, and IFN-ω, which mediate their effects through the interferon-α/β receptor (IFNAR), inducing overlapping yet distinct downstream effects. IFN-I production is triggered by viral infections, double-stranded RNA, and microbial components and can be induced in various leukocyte populations.27-32
Persistent IFN-α overproduction, often described as the IFN-associated gene expression profile, is observed in most paediatric and approximately half present in adults with SLE and correlated with severe involvement of organs, notably the renal and neurological systems. This identifies a subgroup of patients likely to benefit from IFN-targeted therapies.28-30
Previously referred to as natural interferon-producing cells (NIPCs), plasmacytoid dendritic cells function as the principal IFN-α producers. They express CD4, CD36, CD40, CD44, CD45RA, CD83, high CD123, and BDCA-2/4, while lacking CD11c, CD80, and CD86. pDCs display surface and intracellular TLRs, including TLR1, TLR6, TLR7, TLR9, and TLR10, with TLR9 playing a critical role in CpG-DNA–mediated activation. Despite representing approximately making up around 0.1% of circulating mononuclear blood cells, individual pDCs can produce up to 10⁹ IFN-α molecules within 12 hours.30-32
Immunomodulatory Effects of IFN-I Cytokines
IFN-I cytokines exert potent immunostimulatory impact on both innate and adaptive immunity. They enhance Th1 polarization and cellular immune responses driven by cytotoxic T lymphocytes, promote the survival and expansion of memory CD8⁺ T cells, and drive dendritic cell maturation, thereby improving antigen presentation. IFN-α supports B-cell survival, lowers activation thresholds, and induces immunoglobulin class switching via increased levels of BLyS and APRIL. Activated monocytes can function as antigen-presenting cells, further amplifying immune activation. In addition, cytokines plasmacytoid dendritic cells produce cytokines, including IL-12 and IL-15, which stimulate both T-cell and NK-cell responses, collectively contributing to autoimmune pathology in systemic lupus erythematos.27-31
Type I Interferon Pathways in SLE Pathogenesis
SLE affected patient exhibit a marked reduction in circulating plasmacytoid dendritic cells, often exceeding a 70-fold decrease, while residual cells retain robust IFN-α–producing capacity. This reduction likely reflects tissue migration, reinforced by the detection originating from plasmacytoid dendritic cells that produce IFN-α in lupus-affected skin. Endogenous IFN-inducing factors include immune complexes composed of DNA and anti-DNA IgG, which activate pDCs through unmethylated CpG motifs. Apoptotic debris contributes RNA-containing immune complexes that further stimulate IFN-α production, particularly in tissues. Impaired ongoing activation of pDCs is promoted by defective clearance of apoptotic cells and circulating immune complexes, thereby reinforcing autoimmunity.21-28
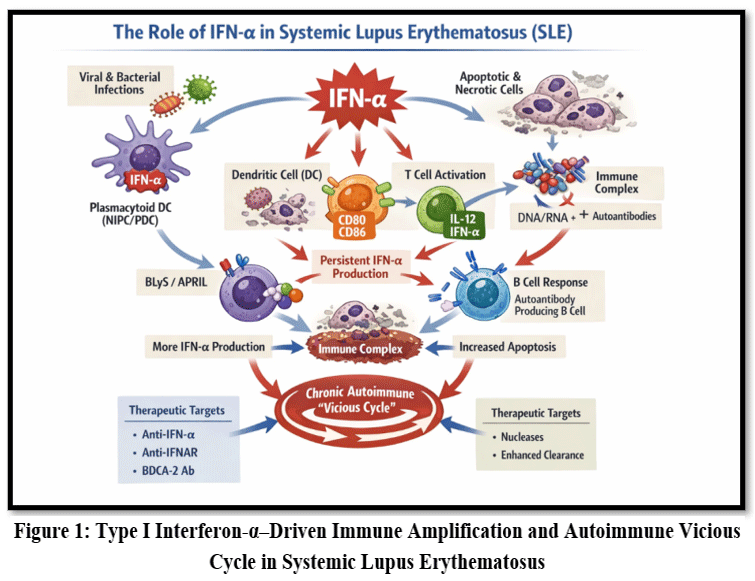 |
Figure 1: Type I Interferon-α–Driven Immune Amplification and Autoimmune Vicious Cycle in Systemic Lupus Erythematosus
|
Sustained IFN-α production promotes dendritic cell maturation, Th1 polarization, and survival of autoreactive B cells. Autoantibodies lead to the formation of immune complexes which boost IFN-α production, resulting in a self-perpetuating inflammatory loop. Regulatory mechanisms, including nucleases, macrophage-mediated clearance, and inhibitory cytokines like IL-10 and TNF-α can terminate this cycle. Disease flares may occur when infections trigger renewed IFN-α release. Therapeutic strategies targeting this pathway include chloroquine, nucleases, FcγRII blockade, anti-IFN-α or anti-IFNAR antibodies, soluble IFNAR, and PDC-directed therapies via BDCA-2.24-32
BAFF and IFNAR1 Signaling
BAFF and IFNAR1 are key immune regulators with distinct biological roles. IFNAR1 is ubiquitously expressed and mediates IFN-I effects by converting local danger signals into systemic inflammation, promoting M1 macrophage polarization, antigen-presenting cell maturation, and antiviral defence. Nearly all nucleated cells express IFNAR1, enabling broad immune activation.5-15
In myeloid cells, PAMPs trigger the activation of pattern recognition receptors, inducing BAFF and IFN-α/β production. Viral nucleic acids are particularly potent inducers of IFN-I. BAFF signals through BAFF-R on transitional, follicular, B cells from the marginal zone and peritoneum, along with dendritic cells and tissue macrophages, thereby supporting adaptive immune responses. BAFF and IFNAR1 signalling jointly promote pro-inflammatory macrophage phenotypes. In contrast, BAFF receptor expression is restricted to specific immune cell subsets. BAFF supports B-cell maturation, survival, antigen presentation, and immunoglobulin secretion, processes essential for immune memory but also central to chronic autoimmunity. Therefore, BAFF inhibition is more selective than IFNAR1 blockade.21-24
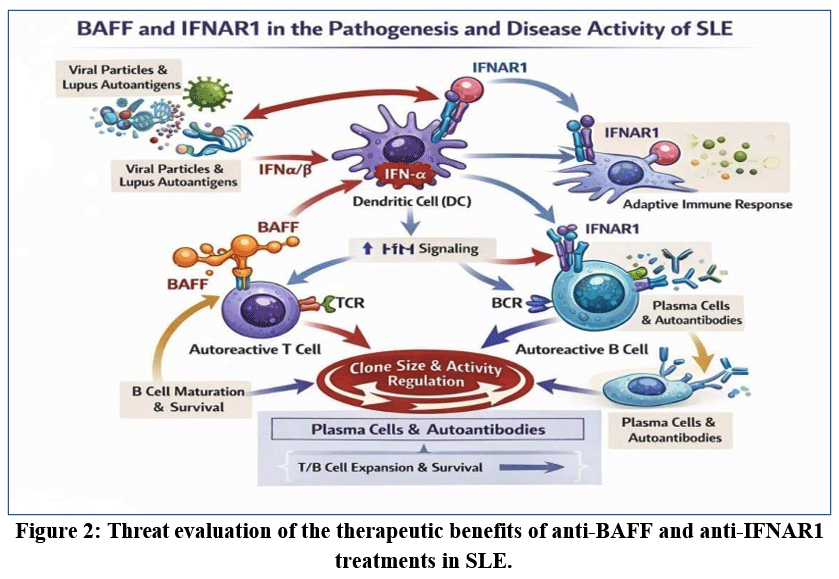 |
Figure 2: Threat evaluation of the therapeutic benefits of anti-BAFF and anti-IFNAR1 treatments in SLE.
|
Infection Risk and Therapeutic Implications
BAFF, together with IFNAR1 contribute to antibacterial and antiviral immunity. BAFF levels increase during bacterial infections and support adaptive immune priming, whereas type I interferons recognise microbial nucleic acids and initiate antiviral defence. Blockade of IFNAR1 poses a greater risk for viral infections compared with BAFF inhibition, reflecting the central role of IFN-I in antiviral immunity.
Belimumab (anti-BAFF), approved in 2011, demonstrates a favourable safety profile, with stable long-term infection rates and rare opportunistic infections. In contrast, anifrolumab (anti-IFNAR1) is effective in individuals experiencing moderate to severe SLE manifestations, particularly in patients with high IFN activity, but is correlated with an elevated risk of herpes zoster as well as additional viral infections. Accordingly, vaccination against herpes zoster and SARS-CoV-2 is advised before the commencement of IFNAR1-targeted therapy.23-32
Table 1: BAFF vs IFNAR1 comparison
| Feature | BAFF | IFNAR1 | Clinical Implication |
| Primary role | B-cell survival | Interferon signalling | Selective vs systemic targeting |
| Expression | Restricted (B cells) | Ubiquitous | Broader immune effect with IFNAR1 |
| Mechanism | Promotes autoantibody production | Amplifies inflammation | Different therapeutic strategies |
| Therapy | Belimumab | Anifrolumab | Depends on IFN signature |
| Infection risk | Lower | Higher (viral infections) | Requires monitoring |
New Understandings and Fresh Viewpoints on Type I Interferon-Targeted Treatment for SLE
This review provides a contemporary and integrative perspective on type I interferon–driven immune dysregulation in systemic lupus erythematosus by directly comparing BAFF- and IFNAR1-mediated pathways as distinct therapeutic targets. Unlike earlier reviews that addressed these mechanisms independently, we emphasize their interrelated roles in shaping innate and adaptive immunity, clinical phenotypes, and therapeutic responsiveness. Importantly, emerging evidence supporting interferon signature–guided patient stratification, therapeutic sequencing, and infection-risk mitigation highlights a transition toward precision-based approaches in SLE management.25
Limitations and Future Directions
Long-term safety gaps: Extended data on infection risk, malignancy, and sustained immunosuppression with interferon-targeted therapies remain limited.
Therapeutic optimization needed: Future studies should assess combination or sequential BAFF and IFNAR1 inhibition to improve efficacy and safety.
Biomarker expansion: The interferon signature alone is insufficient; multi-omic, B-cell, and pDCs-based biomarkers are needed for precision therapy.
Discussion
The IFN-I pathway plays a central role in linking innate immune activation to sustained adaptive autoimmunity in SLE. While BAFF inhibition primarily targets B-cell responses, IFNAR1 blockade suppresses broader inflammatory signaling.
Differences in efficacy and safety highlight the importance of patient stratification using biomarkers such as the interferon signature. Future strategies should focus on combination or sequential therapies and integration of multi-omic biomarkers for precision medicine.
Conclusion
Type I interferon signaling plays a central role in the pathogenesis and progression of systemic lupus erythematosus. Targeting BAFF and IFNAR1 provides promising therapeutic benefits through selective modulation of adaptive and innate immune pathways. Although these biologic therapies improve disease control and reduce flare frequency, careful monitoring for infection-related complications remains essential. Future research should focus on biomarker-guided patient stratification, combination therapeutic approaches, and long-term safety evaluation to support the development of personalized treatment strategies in SLE.
Type I interferon signaling is a key driver of SLE pathogenesis. BAFF and IFNAR1 act as complementary therapeutic targets, each offering distinct advantages.
Future management of SLE will rely on personalized treatment strategies guided by biomarkers, optimized therapy selection, and careful balancing of efficacy with infection risk.
Acknowledgement
The authors would like to acknowledge the Department of Pharmacology, Navsahyadri Institute of Pharmacy, for providing the necessary facilities to conduct this review.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable.
Author Contributions:
Priya Suresh Satav: Conceptualization, Literature Review, Data Collection, Writing–Original Draft Preparation
Kishor Otari: Supervision, Review and Editing, Scientific Guidance
Ajay Kale: Validation, Critical Review, Manuscript Editing
References
- Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2020;383(6):560–571.
- Morand EF, Furie R, Tanaka Y, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. 2020;382(3):211–221.
CrossRef - Furie R, Khamashta M, Merrill JT, et al. Anifrolumab in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. 2019;71(3):393–404.
- Kaul A, Gordon C, Crow MK, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. 2019;5(1):1–21.
- Navarra SV, Guzmán RM, Gallacher AE, et al. Efficacy and safety of belimumab in SLE: long-term extension study. Lupus. 2019;28(10):1231–1240.
- Psarras A, Emery P, Vital EM. Type I interferon-mediated autoimmune diseases. Rheumatology (Oxford). 2019;58(10):1662–1675.
- Crow MK, Olferiev M, Kirou KA. Type I interferons in autoimmune disease. Annu Rev Pathol. 2019;14:369–393.
CrossRef - Kyttaris VC. Type I interferons in systemic lupus erythematosus. J Autoimmun. 2020;108:102398.
- Dörner T, Furie R. Novel paradigms in systemic lupus erythematosus. Lancet. 2019;393(10188):2344–2358.
CrossRef - Muskardin TLW, Niewold TB. Type I interferon in rheumatic diseases. Nat Rev Rheumatol. 2018;14(4):214–228.
CrossRef - Swiecki M, Colonna M. Biology of plasmacytoid dendritic cells. Nat Rev Immunol. 2018;18(7):471–485.
CrossRef - Psarras A, Wittmann M, Vital EM. Plasmacytoid dendritic cells in SLE pathogenesis. Front Immunol. 2020;11:1232.
CrossRef - Rönnblom L, Leonard D. Interferon pathway in SLE. Lupus Sci Med. 2019;6(1):e000270.
CrossRef - Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2018;49(3):397–412.
- Fanouriakis A, Kostopoulou M, Alunno A, et al. EULAR recommendations for SLE management. Ann Rheum Dis. 2019;78(6):736–745.
CrossRef - Hanly JG. Central nervous system lupus. Nat Rev Rheumatol. 2019;15(9):565–579.
- Parikh SV, Almaani S, Brodsky S, Rovin BH. Update on lupus nephritis. J Am Soc Nephrol. 2020;31(3):517–530.
- Touma Z, Gladman DD. Current and future therapies for SLE. Curr Treat Options Rheumatol. 2019;5(3):233–250.
- Wallace DJ, Furie RA, Tanaka Y, et al. Baricitinib for systemic lupus erythematosus. N Engl J Med. 2018;378(26):2467–2478.
- van Vollenhoven RF, Mosca M, Bertsias G, et al. Treat-to-target in SLE. Ann Rheum Dis. 2018;77(7):958–967.
- Rekvig OP. Anti-dsDNA antibodies in SLE. Front Immunol. 2019;10:292.
CrossRef - Harley JB, James JA. Autoantibodies in systemic lupus erythematosus. Curr Opin Immunol. 2020;61:1–7.
- Reed BR, Huff JC, Jones SK, et al. Photosensitivity in lupus erythematosus. Dermatol Clin. 2019;37(3):321–334.
- Rahman A, Isenberg DA. Autoantibodies and clinical correlations in SLE. Rheumatology (Oxford). 2020;59(Suppl 5):v5–v10.
- Klarquist J, Cantrell R, Lehn MA, et al. Type I IFN drives experimental SLE. Immunohorizons. 2020;4(3):140–152.
CrossRef - Tanemura S, Seki N, Tsujimoto H, et al. Role of interferons in Tph cell differentiation. Int Immunol. 2022;34(10):533–544.
CrossRef - Zhao X, Wang S, Wang S, Xie J, Cui D. mTOR signaling in Treg dysfunction in SLE. Clin Immunol. 2022;245:109079.
CrossRef - Sun JL, Lyu TB, Chen ZL, et al. Methylprednisolone and Treg differentiation in SLE. Clin Immunol. 2022;241:109079.
CrossRef - Psarras A, Wittmann M, Vital EM. Type I interferons in SLE pathogenesis and therapy. Nat Rev Rheumatol. 2022;18(10):575–590.
CrossRef - Rönnblom L. Type I interferon system in autoimmunity. Nat Rev Rheumatol. 2022;18(3):133–143.
- Kiefer K, Oropallo MA. Interferon-driven immune dysregulation in SLE. Front Immunol. 2022;13:847712.
- Huang T, Pi C, Xu X, et al. BAFF blockade and B cell receptor repertoire in SLE. Front Immunol. 2024;14:1307392.
CrossRef
Accepted on: 20-06-2026
Second Review by: Dr. Sura Maan Salim
Final Approval by: Dr. Wagih Ghannam








![]()
![]()
