
Immune Regulation of Inflammation: Molecular Signaling, Resolution Mechanisms, and Therapeutic Implications
 , Vaishnavi Vinod Dere, Ajay Yeshawant Kale and Kishor Vasant Otari
, Vaishnavi Vinod Dere, Ajay Yeshawant Kale and Kishor Vasant Otari Department of Pharmacology, Navsahyadri Institute of Pharmacy, Naigaon, India
Corresponding Author E-mail:sushantpatilsp382@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3516
Download this article as:
![]()
Inflammation is an ancient, evolutionarily conserved host-defense response that plays a critical role in protection from infection and injury. Yet, if deregulated, inflammatory responses are a central element in invasive diseases like autoimmune disorders, metabolic syndromes, or cardiovascular and neurodegenerative pathology, and gastrointestinal inflammation. The clinical result of inflammation is not just a product of the extent of immune activation, but rather reflects a balance between pro-inflammatory amplification and active resolution programs. More recently, major advances have utterly changed our thinking on inflammation to that of an active, highly regulated biological process as opposed to a passive cessation of immune signals. This new way of thinking has huge implications for drug discovery. Here, we analyze this recent knowledge, focusing on cellular, molecular and signaling mechanisms controlling immune regulation in the context of inflammation and discuss how activating and regulatory pathways interplay to direct the effects of inflammation. We describe the contribution of innate and adaptive immune cell networks, cytokines, lipid mediators, immune receptors, and intracellular kinases-including NF-κB, MAPK, and JAK/STAT the modulation of inflammatory tone. Crucially, we then reconcile recent insights into immune receptor-mediated regulation and pro-resolving signaling to emphasise why interventions focusing exclusively on inflammatory suppression have demonstrated moderate long-term success. Thinking about inflammation in terms of balance and resolution gives a framework on whichthe next generation of immunomodulatory therapies can be built, specifically designed to promote restoration rather than suppression of immune homeostasis.
KEYWORDS:Cytokines; Immune regulation; Immunopharmacology; Inflammation Resolution biology; Signaling pathways
Introduction
Inflammation is a fundamental and evolutionarily conserved biological response that enables the host to defend against infection, repair tissue injury, and maintain physiological homeostasis. It is initiated through the recognition of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) by pattern recognition receptors (PRRs), leading to activation of innate and adaptive immune responses. Under physiological conditions, inflammation is tightly regulated and self-limiting, resolving once the initiating stimulus has been eliminated.1,11,56
However, dysregulated or unresolved inflammation plays a central role in the pathogenesis of numerous diseases, including autoimmune disorders, metabolic syndrome, cardiovascular diseases, neurodegenerative conditions, cancer, and chronic gastrointestinal disorders. Persistent inflammation is now recognized as a key driver of disease progression and tissue damage across multiple organ systems.2,3,57
Recent advances have shifted the classical view of inflammation from a simple on–off response to a highly dynamic and multi-phase process governed by complex regulatory networks. These networks involve coordinated interactions between immune cells, soluble mediators, and intracellular signaling pathways that determine the magnitude, duration, and outcome of the inflammatory response.60,63
A critical determinant of inflammatory outcome is the balance between pro-inflammatory signaling and active resolution pathways. Pro-inflammatory mediators are essential for host defense; however, failure to appropriately regulate these responses leads to excessive tissue damage and chronic inflammation. In contrast, resolution is now understood as an active and programmed process involving specialized pro-resolving mediators (SPMs), anti-inflammatory cytokines, and cellular clearance mechanisms such as efferocytosis.6,7,58,70
At the molecular level, intracellular signaling pathways such as nuclear factor-kappa B (NF-κB), mitogen-activated protein kinase (MAPK), and Janus kinase/signal transducer and activator of transcription (JAK/STAT) play central roles in integrating inflammatory signals. These pathways regulate gene expression, immune cell activation, and cytokine production, thereby controlling both initiation and resolution of inflammation. Dysregulation of these signaling cascades results in persistent immune activation and contributes to the development of chronic inflammatory diseases.9,24,68,69
In addition to signaling pathways, emerging evidence highlights the importance of immunometabolism and tissue-specific immune regulation in shaping inflammatory responses. Metabolic reprogramming of immune cells influences their functional phenotype, while tissue-specific microenvironments determine the nature and intensity of inflammation. These factors contribute to the heterogeneity observed in inflammatory diseases and represent important considerations for therapeutic targeting.61,62,74
From a pharmacological perspective, traditional anti-inflammatory therapies primarily focus on suppressing inflammatory mediators. Although effective in controlling acute symptoms, these approaches do not restore immune balance and are often associated with significant long-term adverse effects. Consequently, there is increasing interest in therapeutic strategies that promote resolution of inflammation and restore immune homeostasis rather than simply inhibiting immune activation.8,70,71
This review aims to provide a comprehensive overview of the cellular, molecular, and signaling mechanisms underlying immune regulation in inflammation. It emphasizes the concept of inflammatory balance and resolution biology, and discusses emerging therapeutic approaches that target regulatory pathways to achieve sustained and effective control of inflammatory diseases.
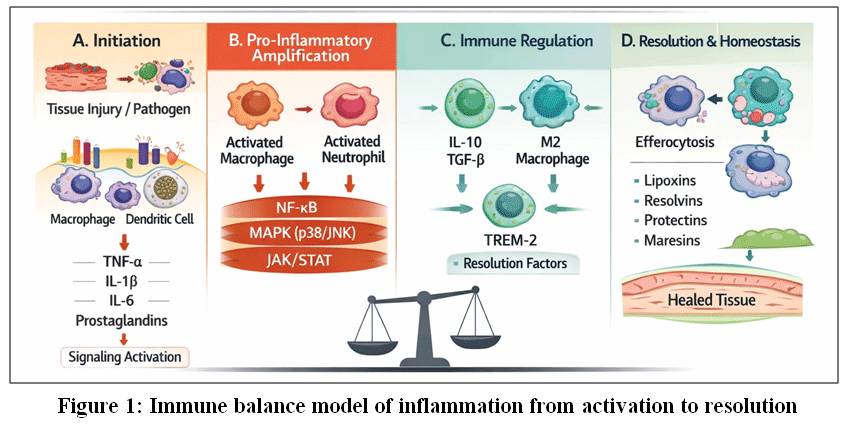 |
Figure 1: Immune balance model of inflammation from activation to resolution
|
This schematic illustrates inflammation as a dynamic and regulated continuum progressing through four interconnected phases: (A) initiation, triggered by tissue injury or pathogen recognition by innate immune cells through pattern-recognition receptors;1,11(B) pro-inflammatory amplification, characterized by activation of macrophages and neutrophils and engagement of NF-κB, MAPK (p38/JNK), and JAK/STAT signaling pathways leading to production of pro-inflammatory cytokines and lipid mediators;2,19,24–27 (C) immune regulation, involving anti-inflammatory cytokines (IL-10, TGF-β), macrophage polarization toward a regulatory phenotype, and receptor-mediated control of inflammatory signaling including TREM-2;4,18,21 and (D) resolution and homeostasis, marked by efferocytosis, generation of specialized pro-resolving mediators (lipoxins, resolvins, protectins, maresins), and restoration of tissue integrity.6,7,30–32 The final inflammatory outcome is determined by the balance between inflammatory activation and active resolution mechanisms.10,37
Novelty and Knowledge Gap
Although several reviews have described inflammatory signaling pathways and immune cell functions, most focus predominantly on pro-inflammatory mechanisms or individual pathways in isolation. A clear integrative framework linking immune activation, immunometabolic regulation, receptor signaling, and resolution biology remains lacking.
This review addresses this gap by
Presenting inflammation as a dynamic balance between activation and resolution, rather than a linear process
Integrating immunometabolism with immune signaling pathways
Highlighting receptor-mediated regulation (TREM axis) as a bridge between innate sensing and resolution
Emphasizing resolution pharmacology as a next-generation therapeutic paradigm
Thus, this review provides a systems-level and mechanistically integrated perspective that extends beyond traditional pathway-based descriptions.
Types and Phases of Inflammation
Inflammation is a fundamental biological response that enables multicellular organisms to protect themselves against infection, tissue injury, and harmful stimuli. Traditionally, inflammation has been classified into acute and chronic forms based on duration, cellular composition, and functional outcomes. However, contemporary immunology recognizes inflammation as a dynamic and continuous process in which acute responses ideally progress toward active resolution, whereas failure of resolution leads to chronic inflammatory pathology.1,2,56 Understanding the distinct phases of inflammation is essential for interpreting both physiological host defense mechanisms and the pathogenesis of inflammatory diseases. These phases are tightly regulated by coordinated interactions between immune cells, molecular mediators, and intracellular signaling pathways.60,63
Acute Inflammation
Acute inflammation represents the immediate and short-term response to tissue injury or infection. It is initiated primarily by tissue-resident immune cells, including macrophages, mast cells, dendritic cells, and innate lymphoid cells, which continuously monitor the microenvironment for danger signals. These cells recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) through pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) and NOD-like receptors (NLRs).11,56
Activation of these receptors triggers intracellular signaling cascades that result in rapid transcriptional and post-transcriptional responses, leading to the release of inflammatory mediators. These include vasoactive amines (e.g., histamine), lipid mediators (prostaglandins and leukotrienes), and pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), which collectively promote vascular changes and immune cell recruitment.12,60
A hallmark of acute inflammation is a well-coordinated vascular response characterized by vasodilation, increased vascular permeability, and leukocyte recruitment. Endothelial activation facilitates leukocyte extravasation through sequential processes of rolling, adhesion, and transmigration mediated by selectins, integrins, and adhesion molecules. Neutrophils are the first responders, followed by monocytes that differentiate into macrophages at the site of inflammation.13–15
Functionally, acute inflammation is protective and promotes pathogen clearance through mechanisms such as phagocytosis, reactive oxygen species (ROS) generation, and enzyme-mediated degradation. Importantly, acute inflammation is self-limiting and is actively resolved through anti-inflammatory cytokines, efferocytosis, and the generation of specialized pro-resolving mediators (SPMs), which restore tissue integrity and immune homeostasis.6,7,58,70
Chronic Inflammation
Chronic inflammation arises when acute inflammatory responses fail to resolve or when persistent stimuli continuously activate the immune system. These stimuli may include unresolved infections, autoimmune reactions, metabolic disturbances, or repeated tissue injury. Under such conditions, sustained immune activation leads to progressive tissue damage and pathological remodeling.3,18,57
Unlike acute inflammation, chronic inflammation is characterized by the prolonged presence of macrophages, lymphocytes, and plasma cells within affected tissues. These cells form complex inflammatory networks that maintain continuous production of cytokines, chemokines, growth factors, and matrix-degrading enzymes, thereby perpetuating inflammation.19–21
A defining feature of chronic inflammation is tissue remodeling, which includes angiogenesis, fibrosis, and extracellular matrix reorganization. While these processes may initially support repair, persistent activation leads to abnormal tissue architecture and organ dysfunction. Sustained fibroblast activation and vascular alterations contribute to irreversible structural damage and disease progression.72
Chronic inflammation is now recognized as a central mechanism underlying a wide range of diseases, including autoimmune disorders, atherosclerosis, metabolic syndrome, neurodegenerative diseases, and inflammatory bowel disease. Despite differences in clinical presentation, these conditions share a common feature: a persistent imbalance between pro-inflammatory and anti-inflammatory signaling pathways.2,57,63
Recent evidence suggests that therapeutic strategies should focus not only on suppressing inflammation but also on restoring immune balance and promoting resolution. Approaches that enhance pro-resolving pathways and regulate immune signaling networks offer improved long-term outcomes compared to conventional anti-inflammatory therapies.70,71
Cellular Components of Inflammation
Inflammatory responses are not mediated by a single cell type but arise from the coordinated and dynamic interactions between immune and non-immune cells within tissues. The cellular composition of inflammation varies depending on the nature of the stimulus, the affected tissue, and the phase of the inflammatory response. During inflammation, immune cells undergo phenotypic and functional reprogramming in response to microenvironmental signals. This cellular plasticity is essential for effective host defense while minimizing tissue damage. Dysregulation of these interactions is a hallmark of chronic inflammatory diseases.23,60,63
Innate Immune Cells
Neutrophils
Neutrophils are the first responders in acute inflammation and are rapidly recruited from the bloodstream to sites of infection or tissue injury. Their migration is guided by chemokines, adhesion molecules, and vascular changes. Upon arrival, neutrophils exert potent antimicrobial effects through phagocytosis, production of reactive oxygen species (ROS), release of proteolytic enzymes, and formation of neutrophil extracellular traps (NETs), which help capture and eliminate pathogens.13,24,25
Although essential for early host defense, excessive or prolonged neutrophil activation can lead to tissue injury. Uncontrolled release of ROS, proteases, and cytotoxic mediators contributes to endothelial damage, extracellular matrix degradation, and amplification of inflammatory signaling. Such dysregulated neutrophil activity is implicated in the transition from acute to chronic inflammation and in the pathogenesis of autoimmune and inflammatory diseases.24,25,56
Macrophages
Macrophages are central regulators of inflammation due to their remarkable functional plasticity and responsiveness to environmental cues. They originate either from circulating monocytes or from tissue-resident populations established during development. Their phenotype and function are shaped by cytokines, microbial products, and metabolic signals present in the local microenvironment.5,59
Traditionally, macrophages have been categorized into pro-inflammatory (M1-like) and anti-inflammatory or reparative (M2-like) phenotypes. M1-like macrophages are induced by microbial stimuli and pro-inflammatory cytokines and produce mediators such as TNF-α, IL-1β, and nitric oxide, thereby enhancing inflammatory responses and microbial clearance. In contrast, M2-like macrophages secrete anti-inflammatory cytokines such as IL-10, promote efferocytosis, and contribute to tissue repair and remodeling.5,26
Macrophage polarization is mechanistically regulated by signaling pathways such as NF-κB and STAT1 in M1 activation, and STAT6 and PPARγ pathways in M2 differentiation, linking extracellular signals to functional phenotypes.
The balance between these functional states is a key determinant of inflammatory outcome. Timely transition from pro-inflammatory to reparative macrophages facilitates resolution and restoration of tissue homeostasis. Conversely, sustained activation of pro-inflammatory macrophages leads to persistent inflammation, fibrosis, and disease progression.59,72
These functional states are further controlled by metabolic reprogramming, where glycolysis supports pro-inflammatory activation, while oxidative phosphorylation promotes anti-inflammatory and pro-resolving functions.
Macrophage Spectrum and Functional Heterogeneity
Recent advances have demonstrated that macrophage activation extends beyond the classical M1/M2 paradigm and instead exists along a dynamic functional spectrum. This spectrum reflects a continuum of activation states influenced by tissue-specific microenvironments, metabolic conditions, and disease context.65
Tissue-resident macrophages exhibit significant heterogeneity depending on their anatomical location. For instance, microglia in the central nervous system, Kupffer cells in the liver, and alveolar macrophages in the lungs possess distinct transcriptional profiles and specialized functions tailored to their respective tissues. This tissue-specific heterogeneity plays a critical role in determining inflammatory responses and disease outcomes.74
Macrophage function is also closely linked to cellular metabolism. Pro-inflammatory macrophages are typically associated with glycolytic metabolism, whereas anti-inflammatory and pro-resolving macrophages rely on oxidative phosphorylation and lipid metabolism. This integration of immunological and metabolic pathways contributes to the diversity of macrophage phenotypes and functions.61,62
Importantly, macrophages exhibit dynamic plasticity and can transition between functional states during the course of inflammation. The ability to shift from a pro-inflammatory to a pro-resolving phenotype is essential for effective resolution and tissue repair. Failure of this transition results in chronic inflammation, impaired resolution, and pathological tissue remodeling.58,70
Understanding macrophage heterogeneity and plasticity is therefore crucial for the development of targeted therapeutic strategies. Approaches aimed at modulating macrophage polarization and enhancing pro-resolving phenotypes hold significant promise for restoring immune balance and treating chronic inflammatory diseases.65,71
Dendritic Cells
Dendritic cells (DCs) serve as a critical link between innate and adaptive immunity. They recognize and process antigens through pattern recognition receptors and present them to naïve T cells, thereby shaping the adaptive immune response. Depending on the cytokine environment and co-stimulatory signals, DCs can promote either inflammatory or regulatory T cell responses.27
In addition to their role in immune activation, DCs are essential for maintaining immune tolerance. Under specific conditions, they induce regulatory T cells and suppress excessive immune responses, thereby preventing tissue damage and chronic inflammation. This dual functionality positions DCs as key regulators of immune balance.60,63
Adaptive Immune Cells
The adaptive immune system plays a major role in chronic inflammation, where prolonged antigen exposure leads to sustained lymphocyte activation. Effector T cells, particularly Th1 and Th17 subsets, promote inflammation by secreting cytokines that activate macrophages and other immune cells. In contrast, regulatory T cells (Tregs) suppress excessive immune responses and contribute to resolution by maintaining immune tolerance.28,66
B cells also contribute to inflammatory processes beyond antibody production. They act as antigen-presenting cells and secrete cytokines that modulate T cell responses and macrophage activation. Dysregulation of B cell function is associated with chronic inflammation and autoimmune diseases.29
Immune Receptors and Regulation
Inflammation is tightly controlled by receptor-mediated signaling mechanisms that regulate immune activation thresholds and response duration. Among these, triggering receptors expressed on myeloid cells (TREM) play important roles in modulating inflammation.
TREM-1 amplifies inflammatory signaling by synergizing with Toll-like receptors, leading to enhanced cytokine production and increased immune activation. Excessive activation of TREM-1 contributes to tissue damage and inflammatory pathology. In contrast, TREM-2 promotes anti-inflammatory responses, enhances phagocytosis, and supports tissue repair, thereby facilitating resolution of inflammation.16–18,75
The balance between activating and inhibitory receptor signaling is a critical determinant of inflammatory outcomes. Disruption of this balance leads to either excessive inflammation or impaired host defense. Therefore, targeting immune receptor signaling represents a promising therapeutic strategy for restoring immune homeostasis in inflammatory diseases.38,75
This figure depicts the contrasting functions of triggering receptors expressed on myeloid cells (TREM) in inflammation. TREM-1, predominantly expressed on neutrophils and macrophages, signals through the adaptor protein DAP12 and SYK kinase to amplify inflammatory responses via NF-κB, MAPK, and JAK/STAT pathways, synergizing with Toll-like receptor signaling and promoting production of pro-inflammatory cytokines, chemokines, and reactive oxygen species, ultimately contributing to tissue injury.16,17,19,24–28 In contrast, TREM-2 signaling on macrophages and dendritic cells limits excessive inflammation by suppressing NF-κB activation, enhancing phagocytosis and efferocytosis, promoting M2 macrophage polarization, and increasing anti-inflammatory cytokine production such as IL-10, thereby facilitating tissue repair and resolution.18,30,31,38 The balance between TREM-1–mediated amplification and TREM-2–mediated regulation critically determines inflammatory outcome and immune homeostasis.10,42
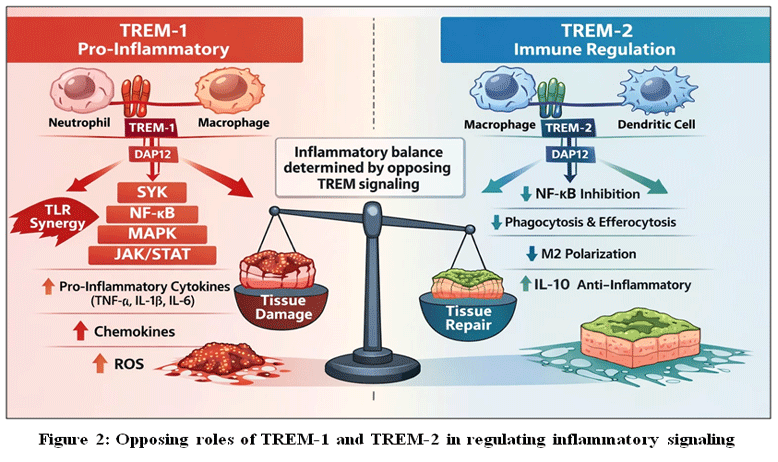 |
Figure 2: Opposing roles of TREM-1 and TREM-2 in regulating inflammatory signaling
|
These cellular interactions are tightly regulated by soluble mediators and intracellular signaling pathways, which are discussed in the following section.
Molecular Mediators of Inflammation
Inflammation is a highly coordinated biological process regulated by a complex network of soluble and cell-associated mediators that control immune cell activation, vascular responses, and tissue repair. These mediators are produced by infiltrating leukocytes, tissue-resident immune cells, endothelial cells, and stromal cells, and act in autocrine and paracrine manners to regulate the initiation, amplification, and resolution of inflammatory responses.19,60
Precise temporal and spatial regulation of these mediators is essential, as excessive or sustained production can lead to tissue injury and chronic inflammatory pathology.2,56
Pro-inflammatory cytokines serve as central regulators of inflammatory signaling. Upon immune activation, cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) are rapidly released and mediate key processes including endothelial activation, leukocyte recruitment, induction of fever, and stimulation of hepatic acute-phase protein synthesis.19–21,66Persistent production of these cytokines promotes amplification of inflammatory cascades and enhances immune cell survival, thereby contributing to chronic inflammation and disease progression.57,66
Chemokines provide specificity to the inflammatory response by directing the migration and positioning of leukocyte subsets at sites of infection or injury. Through the establishment of chemotactic gradients, they ensure efficient immune surveillance and targeted cellular responses, thereby coordinating both innate and adaptive immunity.22,60
Lipid mediators derived from arachidonic acid metabolism, including prostaglandins and leukotrienes, play important roles in regulating vascular tone, increasing vascular permeability, mediating pain, and promoting leukocyte activation. In parallel, reactive oxygen and nitrogen species generated by activated phagocytes contribute to host defense by facilitating microbial killing and amplifying inflammatory signaling. However, excessive production of these reactive species can result in oxidative stress and tissue damage, thereby exacerbating inflammatory pathology.22,23,62
Counter-regulatory mediators are essential for limiting inflammation and promoting its resolution. Anti-inflammatory cytokines such as interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) suppress the production of pro-inflammatory cytokines, inhibit antigen presentation, and maintain immune tolerance.7,21In addition, specialized pro-resolving mediators (SPMs), including lipoxins, resolvins, protectins, and maresins, actively drive the resolution phase by reducing neutrophil recruitment, enhancing efferocytosis, and promoting tissue repair.30,31,58,71
Overall, the outcome of inflammation is determined by the dynamic balance between pro-inflammatory and pro-resolving mediators. Disruption of this balance leads to persistent inflammation and disease, whereas restoration of mediator equilibrium is essential for maintaining immune homeostasis and tissue integrity.32,63
Molecular mediators
Table 1: Key Regulators of Inflammatory Balance
| Component | Pro-inflammatory Role | Resolution Role | Clinical Relevance |
| Cytokines | TNF-α, IL-1β, IL-6 | IL-10, TGF-β | Autoimmune diseases |
| Lipid mediators | Prostaglandins, leukotrienes | Lipoxins, resolvins, protectins | Chronic inflammation |
| Immune cells | Neutrophils, Th1/Th17, M1 macrophages | Tregs, M2 macrophages | Tissue repair |
| Signaling pathways | NF-κB, MAPK, JAK/STAT | Signal termination pathways | Drug targets |
| Immune receptors | TREM-1, TLRs | TREM-2, checkpoint receptors | Precision therapy |
Signaling Pathways in Inflammation
Inflammatory signaling pathways represent central regulatory networks that integrate environmental danger signals into coordinated transcriptional, metabolic, and functional immune responses. These pathways enable immune cells to detect signals from pathogens, tissue injury, and endogenous stress, and respond appropriately to maintain host defense. Among the major intracellular signaling cascades involved in inflammation, the nuclear factor-kappa B (NF-κB), mitogen-activated protein kinase (MAPK), and Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathways are key regulators of inflammatory gene expression, immune cell activation, survival, and effector function.24–29,60
Importantly, these signaling pathways do not operate in isolation but form an interconnected network characterized by extensive crosstalk and feedback regulation. This integration allows precise control over the magnitude, duration, and reversibility of inflammatory responses. While transient activation supports effective host defense, dysregulation of these pathways results in sustained pro-inflammatory signaling and contributes to the development of chronic inflammatory diseases.63,73
NF-κB Signaling Pathway
The NF-κB signaling pathway is a central regulator of inflammation and controls the transcription of numerous genes involved in immune responses, including pro-inflammatory cytokines, chemokines, adhesion molecules, and anti-apoptotic proteins. Through these actions, NF-κB regulates immune cell recruitment, activation, and survival during inflammation.24,25,68
NF-κB activation occurs through phosphorylation and proteasomal degradation of IκB inhibitors, enabling nuclear translocation of NF-κB dimers and transcription of genes encoding pro-inflammatory cytokines, chemokines, adhesion molecules, and anti-apoptotic proteins.
Under physiological conditions, NF-κB activation is rapid and transient. Following immune stimulation, NF-κB translocates to the nucleus, initiates gene transcription, and is subsequently inactivated to prevent excessive immune responses. This tightly controlled activation is essential for effective host defense while limiting tissue damage.
However, persistent or dysregulated NF-κB activation leads to continuous production of inflammatory mediators, resistance to apoptosis, and sustained immune cell activation. Such chronic activation contributes to the pathogenesis of autoimmune diseases, metabolic disorders, cancer, and other inflammatory conditions.68,73
MAPK Signaling Pathways
The MAPK family, including extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinases (JNK), and p38 MAPK, plays a critical role in translating extracellular inflammatory signals into intracellular responses. These pathways regulate both transcriptional and post-transcriptional mechanisms, including cytokine gene expression, mRNA stability, and protein translation.26,27,69
MAPK signaling is mediated through sequential phosphorylation cascades (MAPKKK → MAPKK → MAPK), leading to activation of transcription factors such as AP-1 that regulate cytokine gene expression, cell proliferation, and stress responses.
MAPK pathways are activated by cytokines, microbial components, and cellular stress. Among them, p38 MAPK is particularly important for stabilizing mRNAs encoding inflammatory cytokines such as TNF-α and IL-6, thereby prolonging their expression and amplifying inflammatory responses.
While transient MAPK activation is necessary for appropriate immune responses, sustained activation leads to prolonged inflammatory signaling, cellular stress, and tissue damage. Chronic activation of ERK, JNK, and p38 pathways contributes to persistent inflammation and disease progression.69
JAK/STAT Signaling Pathway
The JAK/STAT pathway serves as a direct link between extracellular cytokine signaling and nuclear gene transcription. Upon cytokine binding to their receptors, Janus kinases (JAKs) are activated, leading to phosphorylation of STAT transcription factors, which subsequently translocate to the nucleus to regulate gene expression.28,29,67
Upon cytokine binding, receptor-associated JAKs phosphorylate specific tyrosine residues, creating docking sites for STAT proteins, which are subsequently phosphorylated, dimerize, and translocate to the nucleus to regulate target gene transcription.
This pathway is essential for immune cell differentiation, activation, and effector functions. However, excessive or prolonged activation of JAK/STAT signaling results in amplification of cytokine signaling loops, sustained immune activation, and immune dysregulation.
Given its central role in cytokine-mediated inflammation, the JAK/STAT pathway has emerged as an important therapeutic target. Pharmacological inhibitors of JAK kinases have demonstrated efficacy in treating inflammatory and autoimmune diseases by modulating aberrant cytokine signaling while preserving essential immune functions.67
Termination of Signaling and Resolution of Inflammation
Effective resolution of inflammation depends on the timely termination of signaling through NF-κB, MAPK, and JAK/STAT pathways. Signal termination mechanisms limit the production of inflammatory mediators, promote resolution pathways, and restore tissue homeostasis.44,70
Failure to appropriately terminate these signaling cascades results in persistent immune activation, continuous cytokine production, and defective resolution of inflammation. Such dysregulation is a key molecular feature of chronic inflammatory and autoimmune diseases.63,73
Restoration of proper signaling regulation and termination has therefore emerged as a critical therapeutic strategy. Targeting these pathways to re-establish immune balance offers promising approaches for the treatment of chronic inflammatory conditions.67,70
Resolution Pathways and Immune Homeostasis
Resolution of inflammation is an active and tightly regulated biological process that is essential for maintaining tissue integrity and immune homeostasis. It is not merely the passive cessation of pro-inflammatory signals but involves coordinated molecular and cellular mechanisms that suppress inflammation and promote tissue repair and immune reprogramming.6,7,58
Anti-inflammatory cytokines such as interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) play crucial roles in limiting inflammatory responses. IL-10 inhibits the activation of macrophages and dendritic cells, whereas TGF-β promotes immune tolerance and tissue repair. Dysregulation of these cytokines leads to persistent inflammation, fibrosis, and impaired resolution.21,57
Specialized pro-resolving mediators (SPMs), including lipoxins, resolvins, protectins, and maresins, represent a major advancement in resolution biology. Derived from omega-3 and omega-6 fatty acids, these mediators actively terminate inflammation by reducing neutrophil recruitment, enhancing efferocytosis, and promoting tissue regeneration while preserving host defense mechanisms.30,31,58,71
Efferocytosis, the clearance of apoptotic cells by phagocytes, is a critical process in inflammation resolution. Efficient efferocytosis suppresses pro-inflammatory signaling and promotes the release of anti-inflammatory mediators. In contrast, defective clearance leads to secondary necrosis, sustained immune activation, and chronic inflammation.32,70
Collectively, cytokines, lipid mediators, and cellular clearance pathways form an integrated resolution network that restores immune balance and tissue homeostasis. Disruption of this network is a key contributor to chronic inflammatory diseases.63
Immunometabolism in Inflammatory Regulation
Immunometabolism refers to the interplay between cellular metabolic pathways and immune cell function. During inflammation, immune cells undergo metabolic reprogramming that directly influences their phenotype and effector functions.
Pro-inflammatory immune cells, such as M1 macrophages, rely predominantly on aerobic glycolysis (Warburg-like metabolism), which supports rapid ATP generation and biosynthesis of inflammatory mediators. This metabolic state promotes activation of NF-κB and production of cytokines such as TNF-α and IL-1β.
In contrast, anti-inflammatory and pro-resolving macrophages (M2-like) depend on oxidative phosphorylation and fatty acid oxidation, which support tissue repair and resolution processes.
Key metabolic intermediates such as succinate, citrate, and itaconate act as signaling molecules that regulate inflammatory pathways. For example:
Succinate stabilizes HIF-1α → promotes IL-1β
Itaconate inhibits NF-κB → anti-inflammatory effect
Additionally, metabolic pathways regulate:
Epigenetic modifications
Cytokine production
Immune cell differentiation
Thus, immunometabolism represents a critical mechanistic link between cellular metabolism and immune regulation, offering novel therapeutic targets for controlling inflammation.
Pharmacological Modulation of Inflammatory Balance
Pharmacological modulation of inflammation has traditionally focused on suppressing excessive immune responses to alleviate symptoms and limit tissue damage. Conventional therapeutic strategies primarily target the production or activity of pro-inflammatory mediators rather than restoring immune homeostasis. Although effective in managing acute inflammation, these approaches often fail to address the underlying imbalance in chronic inflammatory diseases.33,34,56
Classical Anti-Inflammatory Therapies
Nonsteroidal anti-inflammatory drugs (NSAIDs) and glucocorticoids remain the cornerstone of anti-inflammatory treatment. NSAIDs inhibit cyclooxygenase enzymes, thereby reducing prostaglandin synthesis, while glucocorticoids suppress multiple inflammatory pathways through transcriptional regulation of pro-inflammatory genes.
Despite their effectiveness, these agents do not actively promote resolution of inflammation. Long-term or high-dose use is associated with significant adverse effects, including gastrointestinal toxicity, metabolic disturbances, osteoporosis, adrenal suppression, and increased susceptibility to infections. These limitations highlight the drawbacks of broad immunosuppression in achieving sustained immune balance.33,34,56
Targeted Immunomodulatory Therapies
Advances in molecular immunology have led to the development of targeted therapies that selectively inhibit specific inflammatory mediators and signaling pathways. Biological agents targeting TNF-α, IL-6, and IL-1β have significantly improved the management of autoimmune and chronic inflammatory diseases by providing pathway-specific inhibition.35,66
In addition, small-molecule Janus kinase (JAK) inhibitors have emerged as effective oral therapies that modulate intracellular cytokine signaling. These agents offer controlled and reversible immunomodulation, allowing partial suppression of inflammatory pathways while preserving essential immune functions.36,37,67
Limitations of Suppressive Strategies
Despite therapeutic advancements, most current anti-inflammatory treatments are fundamentally suppressive in nature. By broadly reducing immune activity, these therapies may compromise host defense and increase susceptibility to infections and malignancies. Importantly, they do not correct the imbalance between pro-inflammatory and regulatory pathways that underlies chronic inflammation.
As a result, disease recurrence is common after treatment discontinuation, indicating that suppressive strategies fail to achieve long-term immune homeostasis.56,63
Resolution-Based Therapeutic Approaches
Emerging therapeutic strategies focus on promoting resolution of inflammation rather than suppressing immune activation. These approaches aim to enhance endogenous anti-inflammatory and pro-resolving pathways, particularly through the modulation of specialized pro-resolving mediators (SPMs) and their signaling mechanisms.58,70,71
Unlike conventional therapies, resolution-based approaches actively terminate inflammation, promote clearance of inflammatory cells, and restore tissue integrity while preserving immune competence. This represents a paradigm shift toward restoring immune balance rather than inhibiting immune responses.70,71
Conceptual Shift in Inflammatory Pharmacology
Overall, these developments reflect a major shift in inflammatory pharmacology—from broad immune suppression to selective modulation of immune balance and resolution pathways. Future therapies will increasingly focus on regulating the intensity and duration of inflammation, promoting timely resolution, and maintaining immune homeostasis.
This evolving paradigm offers the potential for more effective and safer therapeutic strategies, enabling long-term disease control with minimal adverse effects and advancing the development of next-generation anti-inflammatory treatments.70,73
Figure 3. Contrasting pharmacological strategies: inflammatory suppression versus resolution-based therapy
This illustration compares conventional inflammatory suppression therapies with emerging resolution-based pharmacological approaches. Traditional anti-inflammatory treatments, including non-steroidal anti-inflammatory drugs, corticosteroids, and anti-cytokine biologics, primarily act by inhibiting cyclooxygenase activity and suppressing NF-κB/MAPK signaling, resulting in reduced cytokine production but often causing broad immune suppression, impaired host defense, delayed tissue healing, and increased risk of relapse.8,33–35 In contrast, resolution-based therapies aim to promote endogenous termination of inflammation through specialized pro-resolving mediators (lipoxins, resolvins, protectins, maresins), activation of regulatory receptors, enhancement of efferocytosis, and immune reprogramming toward homeostasis.7,30–32,37 These strategies support durable tissue repair and immune balance without compromising antimicrobial defense.6,10,41
Clinical Implications and Future Perspectives
Dysregulated inflammation has emerged as a central contributor to a wide spectrum of acute and chronic diseases, including autoimmune disorders, cardiovascular diseases, metabolic syndrome, neurodegenerative conditions, and inflammatory bowel disease.39–41,57 In these conditions, inflammation is not merely a secondary consequence of tissue injury but actively participates in disease initiation, progression, and complication. Persistent inflammatory signaling drives tissue damage, metabolic dysfunction, and immune imbalance, thereby exacerbating disease severity.3,63
Importantly, complete elimination of inflammation is neither biologically feasible nor clinically desirable. Inflammatory responses are essential for host defense, tissue repair, and immune surveillance. Therefore, therapeutic strategies aimed at total suppression of inflammation may disrupt critical physiological processes and increase susceptibility to infections and other complications.56,70
Current trends in inflammatory medicine emphasize controlled immune modulation rather than broad immunosuppression. The goal is to restore immune balance and re-establish homeostasis by fine-tuning inflammatory responses rather than completely inhibiting them. This approach allows preservation of protective immune functions while preventing excessive tissue damage and chronic disease progression.63,71
Emerging evidence highlights the importance of personalized and precision-based therapeutic strategies that consider tissue-specific inflammatory mechanisms, immune cell heterogeneity, and patient-specific factors. Advances in systems immunology, immunometabolism, and molecular profiling are enabling the identification of novel therapeutic targets and biomarkers for disease stratification and treatment optimization.61,62,64
Future therapeutic approaches are expected to focus on enhancing endogenous resolution pathways, modulating immune receptor signaling, and targeting key intracellular signaling networks. Resolution-based therapies, including those targeting specialized pro-resolving mediators (SPMs), represent a promising direction for achieving sustained disease control with minimal adverse effects.58,70,71
Overall, a paradigm shift is underway in inflammatory disease management—from symptom suppression to restoration of immune balance. This evolving framework offers the potential for more effective, targeted, and safer therapeutic strategies, ultimately improving long-term clinical outcomes and patient quality of life.70,73
Implications of Targeted Immunotherapies
The emergence of pathway-specific immunotherapies has demonstrated that selective inhibition of key inflammatory drivers can provide substantial therapeutic benefits. By targeting specific signaling pathways and cytokine networks, these therapies can disrupt disease-driving mechanisms while minimizing systemic toxicity. Such targeted approaches have transformed the management of several inflammatory and immune-mediated diseases by modulating the inflammatory milieu rather than indiscriminately suppressing immune function.35,66,67
Despite these advances, important limitations remain. Inter-individual variability in immune responses, disease heterogeneity, and tissue-specific differences in inflammatory networks contribute to variability in therapeutic outcomes. In addition, concerns regarding long-term safety, immunogenicity, and sustained immune modulation persist, particularly with biologics and advanced cellular therapies. These challenges highlight the limitations of “one-size-fits-all” treatment strategies and underscore the need for personalized therapeutic approaches that consider individual immune balance, resolution capacity, and tissue-specific regulation.42,43,64
Toward Personalized Inflammatory Medicine
Future management of inflammatory diseases is expected to integrate advances in systems immunology, biomarker discovery, and precision pharmacology. Systems-level approaches enable comprehensive mapping of immune networks and signaling pathways, providing deeper insights into the regulation of inflammatory responses.61,63
Identification of reliable biomarkers reflecting inflammatory burden, resolution capacity, and pathway activation is critical for patient stratification and treatment optimization. Such biomarkers can guide selection of appropriate therapies, predict treatment response, and allow dynamic monitoring of disease progression. Personalized immunomodulation strategies based on biomarker-driven decision-making have the potential to enhance therapeutic efficacy, reduce adverse effects, and achieve sustained disease control.44–47,64
In parallel, modulation of immune checkpoints beyond oncology is emerging as a novel strategy for regulating inflammation. By adjusting immune activation thresholds rather than inducing global immunosuppression, checkpoint-based approaches can provide precise control over inflammatory responses while preserving essential immune functions.55,75
Translational Perspective and Therapeutic Long-Term Goals
From a translational perspective, therapies that enhance endogenous resolution pathways, restore immune receptor balance, and normalize tissue microenvironments represent a transformative approach to inflammatory disease management. Unlike conventional anti-inflammatory therapies that broadly inhibit immune responses, these strategies align pharmacological intervention with natural regulatory mechanisms of the immune system.58,70,71
Restoration of immune homeostasis has both short- and long-term clinical implications. In the short term, such approaches may reduce disease flares, improve symptom control, and minimize treatment-related toxicity. Over the long term, they hold the potential to slow disease progression, improve survival outcomes, and reduce dependence on chronic pharmacotherapy.
Ultimately, the induction and maintenance of physiological immune regulation represent a rational and sustainable strategy for managing inflammatory diseases. This paradigm shift—from suppression to restoration of immune balance—offers a promising foundation for the development of next-generation therapeutic interventions.48–55,73
Discussion
The present review highlights inflammation as a highly dynamic and tightly regulated process governed by complex interactions between immune cells, molecular mediators, and intracellular signaling pathways. While significant progress has been made in understanding inflammatory mechanisms, several important aspects require further consideration to fully explain the variability and complexity observed in inflammatory diseases.
A central concept emerging from recent research is that inflammation cannot be adequately explained by linear pathways alone. Instead, it represents a multidimensional network in which signaling pathways such as NF-κB, MAPK, and JAK/STAT interact through extensive crosstalk and feedback mechanisms. This network-based regulation allows precise control of inflammatory responses but also introduces multiple points at which dysregulation can occur, leading to chronic inflammation and disease progression.60,63,73
One of the major limitations of traditional models is the oversimplified classification of immune cells, particularly macrophages. The classical M1/M2 paradigm does not fully capture the functional diversity of macrophages observed in vivo. Emerging evidence supports the concept of a macrophage activation spectrum, in which cells adopt context-dependent phenotypes influenced by tissue microenvironment, metabolic state, and disease conditions.65This spectrum-based model provides a more accurate framework for understanding how macrophages contribute to both inflammation and resolution.
In addition to cellular diversity, tissue-specific inflammatory heterogeneity represents a critical but often underappreciated factor. Inflammatory responses differ significantly across tissues due to variations in immune cell composition, stromal interactions, metabolic environment, and local signaling networks. For example, microglia in the central nervous system, Kupffer cells in the liver, and adipose tissue macrophages exhibit distinct functional profiles that influence disease outcomes in neurodegeneration, liver disease, and metabolic disorders, respectively.61,62,74These differences suggest that inflammation is not a uniform process but rather a context-dependent phenomenon requiring tissue-specific therapeutic approaches.
Another important consideration is the balance between pro-inflammatory and pro-resolving mechanisms. While most current therapeutic strategies focus on suppressing inflammatory mediators, increasing evidence indicates that failure of resolution pathways is a key driver of chronic inflammation. Defects in specialized pro-resolving mediators (SPMs), impaired efferocytosis, and dysregulated anti-inflammatory cytokine signaling contribute to persistent inflammation and tissue damage.58,70,71Therefore, inflammation should be viewed not only as excessive activation but also as defective resolution.
Alternative hypotheses further suggest that metabolic and environmental factors play a crucial role in shaping inflammatory responses. Immunometabolism has emerged as a key regulator of immune cell function, influencing macrophage polarization, cytokine production, and signaling pathway activation. Metabolic reprogramming can either promote inflammatory responses or support resolution, depending on the context.61,62 Similarly, trained immunity and epigenetic reprogramming may contribute to sustained inflammatory responses even after removal of the initial stimulus, providing an additional layer of complexity.64
From a therapeutic perspective, these insights challenge the traditional paradigm of broad immunosuppression. While targeted therapies against cytokines and signaling pathways have improved clinical outcomes, they do not fully address the underlying dysregulation of immune balance. Inter-individual variability, tissue-specific responses, and network-level interactions limit the effectiveness of uniform treatment strategies.66,67
Taken together, these observations support a shift toward a systems-level understanding of inflammation that integrates cellular heterogeneity, tissue-specific context, metabolic regulation, and resolution biology. Future research should focus on identifying precise biomarkers, understanding tissue-specific immune networks, and developing therapies that restore immune balance rather than simply suppress inflammatory pathways. Such approaches are likely to provide more effective and sustainable strategies for the management of inflammatory diseases.63,70,73
Importantly, this review integrates emerging concepts such as immunometabolism, macrophage heterogeneity, and resolution pharmacology into a unified framework of inflammatory regulation. This integrative perspective highlights potential therapeutic strategies aimed at restoring immune balance rather than suppressing immune responses.
Conclusion
Inflammation is an evolutionarily conserved biological response essential for host defense, tissue repair, and immune surveillance. However, its protective function depends on precise regulation of both the intensity and duration of the response. As discussed, inflammation is not a simple on–off phenomenon but a dynamic and tightly coordinated process governed by complex interactions among cellular, molecular, and signaling networks. Importantly, the transition from acute, protective inflammation to chronic disease is driven not only by sustained activation but also by failure of regulatory and resolution mechanisms.
Recent advances have established that resolution of inflammation is an active and highly regulated process involving coordinated actions of anti-inflammatory cytokines, pro-resolving mediators, immune regulatory receptors, and cellular clearance pathways. Conversely, persistent activation of key signaling pathways such as NF-κB, MAPK, and JAK/STAT contributes to immune dysregulation, tissue injury, and progression of chronic inflammatory diseases. These insights highlight the importance of maintaining a balance between activation and resolution to preserve immune homeostasis.
From a therapeutic perspective, current evidence indicates that conventional anti-inflammatory strategies based primarily on broad immune suppression are effective for short-term symptom control but are limited in restoring long-term immune balance. These approaches may also be associated with adverse effects due to non-specific inhibition of immune function.
Emerging therapeutic strategies are therefore shifting toward targeted immunomodulation and enhancement of endogenous resolution pathways. Approaches such as selective cytokine inhibition, modulation of intracellular signaling pathways, and promotion of resolution mechanisms offer the potential to achieve more controlled and sustained regulation of inflammation. In addition, advances in systems immunology and biomarker-guided therapy are expected to support more individualized treatment strategies that account for variability in immune responses and disease context.
Overall, improving our understanding of inflammatory regulation and resolution will be critical for the development of safer and more effective therapeutic interventions. A balanced approach that integrates controlled immune modulation with preservation of host defense represents a rational and sustainable strategy for managing inflammatory diseases.
Acknowledgement
The authors would like to acknowledge the academic support and constructive suggestions provided by colleagues and mentors during the preparation of this review.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Author Contributions
- Sushant Satappa Patil: Conceptualization, literature review, data collection, writing – original draft preparation, and manuscript organization.
- Vaishnavi Vinod Dere: Literature survey, data compilation, reference management, and writing – review & editing.
- Ajay Yeshawant Kale: Scientific guidance, methodology development, critical revision of the manuscript, and supervision.
- Kishor Vasant Otari: Project administration, overall supervision, final review, and approval of the manuscript for publication.
References
- Medzhitov R. Origin and physiological roles of inflammation. 2008;454(7203):428-435. doi:10.1038/nature07201
CrossRef - Nathan C, Ding A. Nonresolving inflammation. 2010;140(6):871-882. doi:10.1016/j.cell.2010.02.029
CrossRef - Furman D, Campisi J, Verdin E, et al. Chronic inflammation in the etiology of disease. Nat Med. 2019;25(12):1822-1832. doi:10.1038/s41591-019-0675-0
CrossRef - O’Garra A, Vieira P. Regulatory T cells and mechanisms of immune regulation. Nat Med. 2004;10(8):801-805. doi:10.1038/nm0804-801
CrossRef - Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723-737. doi:10.1038/nri3073
CrossRef - Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6(12):1191-1197. doi:10.1038/ni1276
CrossRef - Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. 2014;510(7503):92-101. doi:10.1038/nature13479
CrossRef - Barnes PJ. How corticosteroids control inflammation. Br J Pharmacol. 2006;148(3):245-254. doi:10.1038/sj.bjp.0706736
CrossRef - O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. 2012;36(4):542-550. doi:10.1016/j.immuni.2012.03.014
CrossRef - Schett G, Neurath MF. Resolution of chronic inflammatory disease. Nat Commun. 2018;9:3261. doi:10.1038/s41467-018-05800-6
CrossRef - Takeuchi O, Akira S. Pattern recognition receptors and inflammation. 2010;140(6):805-820. doi:10.1016/j.cell.2010.01.022
CrossRef - Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. 2014;41(5):694-707. doi:10.1016/j.immuni.2014.10.008
CrossRef - Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6(3):173-182. doi:10.1038/nri1785
CrossRef - Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214(2):199-210. doi:10.1002/path.2277
CrossRef - Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. 10th ed. Philadelphia, PA: Elsevier; 2021.
- Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol. 2003;3(6):445-453. doi:10.1038/nri1106
CrossRef - Bouchon A, Dietrich J, Colonna M. Inflammatory responses can be triggered by TREM-1. J Immunol. 2000;164(10):4991-4995. doi:10.4049/jimmunol.164.10.4991
CrossRef - Ulland TK, Colonna M. TREM2—a key player in microglial biology and inflammatory regulation. Nat Rev Neurol. 2018;14(11):667-675. doi:10.1038/s41582-018-0072-1
CrossRef - Dinarello CA. Proinflammatory cytokines. 2000;118(2):503-508. doi:10.1378/chest.118.2.503
CrossRef - Kishimoto T. IL-6: from laboratory to bedside. Clin Rev Allergy Immunol. 2005;28(3):177-186. doi:10.1385/CRIAI:28:3:177
CrossRef - Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10(3):170-181. doi:10.1038/nri2711
CrossRef - Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986-1000. doi:10.1161/ATVBAHA.110.207449
CrossRef - Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20(7):1126-1167. doi:10.1089/ars.2012.5149
CrossRef - Lawrence T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi:10.1101/cshperspect.a001651
CrossRef - Hayden MS, Ghosh S. NF-κB in immunobiology. Cell Res. 2011;21(2):223-244. doi:10.1038/cr.2011.13
CrossRef - Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55-72. doi:10.1146/annurev.immunol.20.091301.131133
CrossRef - Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13(9):679-692. doi:10.1038/nri3495
CrossRef - Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;16(12):843-862. doi:10.1038/nrd.2017.201
CrossRef - Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK–STAT signaling as a target for inflammatory disease therapy. Int J Mol Sci. 2017;18(11):2329. doi:10.3390/ijms18112329
CrossRef - Buckley CD, Gilroy DW, Serhan CN. Immune regulation by apoptotic cell clearance. Nat Rev Immunol. 2011;11(1):59-69. doi:10.1038/nri2932
CrossRef - Fredman G, Serhan CN. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Cell Mol Life Sci. 2011;68(12):1997-2014. doi:10.1007/s00018-011-0709-z
CrossRef - Elliott MR, Ravichandran KS. The dynamics of apoptotic cell clearance. Dev Cell. 2016;38(2):147-160. doi:10.1016/j.devcel.2016.06.029
CrossRef - Vane JR, Botting RM. Mechanism of action of anti-inflammatory drugs. Am J Med. 1998;104(3A):2S-8S. doi:10.1016/S0002-9343(97)00203-9
CrossRef - Schäcke H, Döcke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96(1):23-43. doi:10.1016/S0163-7258(02)00297-8
CrossRef - Feldmann M, Maini RN. Anti-TNF therapy, from rationale to standard of care. Nat Med. 2003;9(10):1245-1250. doi:10.1038/nm939
CrossRef - O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. JAK inhibition in autoimmune diseases. Nat Rev Drug Discov. 2017;16(12):843-862. doi:10.1038/nrd.2017.201
CrossRef - Serhan CN. Resolution pharmacology: pro-resolving mediators in inflammation. Nat Rev Drug Discov. 2014;13(1):79-98. doi:10.1038/nrd4135
CrossRef - Klesney-Tait J, Turnbull IR, Colonna M. The TREM receptor family and signal integration. Nat Immunol. 2006;7(12):1266-1273. doi:10.1038/ni1411
CrossRef - Smolen JS, Aletaha D, Bijlsma JWJ, et al. Treating rheumatoid arthritis to target. Ann Rheum Dis. 2010;69(4):631-637. doi:10.1136/ard.2009.123919
CrossRef - Firestein GS, McInnes IB. Immunopathogenesis of rheumatoid arthritis. 2017;46(2):183-196. doi:10.1016/j.immuni.2017.02.006
CrossRef - Schett G. Resolution of inflammation in arthritis. Nat Rev Rheumatol. 2019;15(2):83-96. doi:10.1038/s41584-018-0139-2
CrossRef - Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. 2012;335(6071):936-941. doi:10.1126/science.1214935
CrossRef - Nathan C. Points of control in inflammation. 2002;420(6917):846-852. doi:10.1038/nature01320
CrossRef - Kawai T, Akira S. Regulation of innate immune signalling pathways. Nat Rev Immunol. 2011;11(3):173-184. doi:10.1038/nri2950
CrossRef - Delhalle S, Blasius R, Dicato M, Diederich M. NF-κB-dependent gene expression in immune responses. Biochem Pharmacol. 2004;68(1):1-14. doi:10.1016/j.bcp.2004.03.011
CrossRef - Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Nat Rev Immunol. 2014;14(11):706-718. doi:10.1038/nri3749
CrossRef - Hotamisligil GS. Inflammation and metabolic disorders. 2006;444(7121):860-867. doi:10.1038/nature05485
CrossRef - Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. 2013;339(6116):166-172. doi:10.1126/science.1230720
CrossRef - Libby P. Inflammation in atherosclerosis. 2002;420(6917):868-874. doi:10.1038/nature01323
CrossRef - Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. 2011;144(5):646-674. doi:10.1016/j.cell.2011.02.013
CrossRef - Prinz M, Priller J. The role of neuroinflammation in neurodegenerative diseases. Nat Rev Neurosci. 2014;15(5):300-312. doi:10.1038/nrn3723
CrossRef - Calder PC. Omega-3 fatty acids and inflammatory processes. 2010;2(3):355-374. doi:10.3390/nu2030355
CrossRef - Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. 2008;372(9655):756-765. doi:10.1016/S0140-6736(08)61039-9
CrossRef - Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Nat Neurosci. 2016;19(8):987-995. doi:10.1038/nn.4307
CrossRef - Vignali DAA, DePeaux K, Watson MJ, Ye C. Immune checkpoints beyond cancer. Nat Rev Immunol. 2018;18(9):537-548. doi:10.1038/s41577-018-0006-4
- Chen L, Deng H, Cui H, et al. Inflammatory responses and inflammation-associated diseases in organs. 2022;13:1234-1250.
- Nathan C. The resolution of inflammation: principles and challenges. Nat Rev Immunol. 2023;23:123-135.
- Serhan CN. Pro-resolving mediators in inflammation and disease. 2023;615:45-56.
- Wynn TA, Vannella KM. Macrophage biology in development and disease. Nat Rev Immunol. 2023;23:345-362.
- Glass CK, Natoli G. Molecular control of inflammation. Nat Immunol. 2022;23:789-798.
- O’Neill LAJ, Artyomov MN. Itaconate and immune metabolism in inflammation. 2022;185:1559-1573.
- Hotamisligil GS. Inflammation, metabolism and immunometabolism. 2022;602:339-349.
- Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. 2023;186:456-472.
- Mantovani A, Netea MG. Trained immunity and inflammation. 2023;379:eabo1234.
- Murray PJ. Macrophage polarization: beyond M1/M2. Nat Rev Immunol. 2024;24:123-138.
- Schett G, McInnes IB. Cytokine networks in inflammatory disease. Nat Med. 2023;29:456-468.
- Banerjee S, Biehl A. Advances in JAK-STAT signaling in inflammation. Nat Rev Drug Discov. 2024;23:567-580.
CrossRef - Lawrence T. NF-κB signaling in inflammation and disease. Nat Rev Immunol. 2023;23:89-102.
- Arthur JSC. MAPK signaling in immune responses. Nat Rev Immunol. 2023;23:210-224.
- Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. 2022;21:567-584.
- Chiang N, Serhan CN. Specialized pro-resolving mediators in immunity. Nat Rev Immunol. 2023;23:381-395.
- Wynn TA. Fibrosis and chronic inflammation. Nat Rev Immunol. 2022;22:333-346.
CrossRef - Medzhitov R. Origin and functions of inflammatory responses. 2024;620:45-56.
- Orecchioni M, et al. Tissue-specific macrophage heterogeneity. Nat Immunol. 2023;24:234-245.
- Vignali DAA. Immune checkpoints in inflammation. Nat Rev Immunol. 2023;23:567-582.
Abbreviations
NF-κB: Nuclear Factor kappa B
MAPK: Mitogen Activated Protein Kinase
JAK: Janus Kinase
STAT: Signal Transducer and Activator of Transcription
TNF-α: Tumor Necrosis Factor Alpha
IL: Interleukin
SPMs: Specialized Pro-Resolving Mediators
TLRs: Toll Like Receptors
PRRs: Pattern Recognition Receptors
ROS: Reactive Oxygen Species
Accepted on: 16-04-2026
Second Review by: Dr. Arnaw Kishore
Final Approval by: Dr. Jagdish Chandra Joshi








![]()
![]()
