
Gene Therapy and CRISPR-based Intervention in Sickle Cell Anemia
 , Sushant Satappa Patil, Ajay Yeshawant Kale and Kishor Vasant Otari
, Sushant Satappa Patil, Ajay Yeshawant Kale and Kishor Vasant Otari Department of Pharmacology, Navsahyadri Institute of Pharmacy, Naigaon, India
Corresponding Author Email:vaishnavidere17@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3515
Download this article as:
![]()
Sickle cell anemia is a severe inherited blood disorder caused by a mutation in the beta globin gene, leading to the formation of abnormal hemoglobin S and deformation of red blood cells into rigid, sickle-shaped structures. These altered cells impair blood flow within the microcirculation, resulting in recurrent vaso-occlusive episodes, chronic inflammation, progressive organ damage, and reduced quality of life. Conventional therapeutic approaches, including hydroxyurea therapy, repeated blood transfusions, and hematopoietic stem cell transplantation, can reduce disease severity but do not correct the underlying genetic defect.The objective of this review is to provide a comprehensive evaluation of gene therapy strategies, with particular emphasis on clustered regularly interspaced short palindromic repeats and CRISPR associated protein 9 based genome editing, in the treatment of sickle cell disease, focusing on their mechanisms, clinical outcomes, and associated challenges.Recent advances in gene editing enable targeted correction of the mutated beta globin gene or reactivation of fetal hemoglobin through modification of regulatory elements such as B cell lymphoma eleven A. The first approved gene editing therapy, Casgevy, has demonstrated substantial clinical benefits, including a marked reduction in vaso-occlusive episodes and improved transfusion independence.Despite these promising outcomes, several challenges remain, including unintended genomic modifications, toxicity associated with conditioning regimens, high treatment costs, limited accessibility, and ethical concerns related to genome integrity. Continued advancements in technology, ethical oversight, and healthcare policy are essential to ensure safe, effective, and equitable clinical implementation of gene therapy as a potentially curative approach for sickle cell disease.
KEYWORDS:CRISPR/Cas9; Gene Therapy; Genome Editing; Hemoglobin; Sickle Cell Disease
Introduction
Gene therapy is an innovative therapeutic strategy for the treatment of genetic diseases at the molecular level, rather than merely alleviating clinical manifestations.¹ Gene therapy utilizes advanced molecular tools, including systems such as clustered regularly interspaced short palindromic repeats and CRISPR associated protein 9, to enable precise correction of defective genes and offers the potential for long-term or even permanent therapeutic benefit.² A significant breakthrough in this field was the approval of Casgevy, a genome editing-based therapy approved by the United States Food and Drug Administration for the treatment of sickle cell disease.³⁻⁵This approach involves the modification of autologous hematopoietic stem cells to induce the expression of fetal hemoglobin, which effectively inhibits the polymerization of sickle hemoglobin and prevents red blood cell sickling.⁶ Clinical trial outcomes have demonstrated a substantial reduction in painful vaso-occlusive episodes, confirming its therapeutic efficacy. With approval for patients aged twelve years and above, Casgevy represents a potentially curative, single-administration treatment.
The therapeutic significance of gene therapy lies in its ability to correct the underlying genetic defect, reduce dependence on lifelong supportive care, and provide a safer alternative to allogeneic bone marrow transplantation. Consequently, gene-based therapies can significantly improve patient quality of life by reducing transfusion requirements, decreasing hospitalization frequency, and minimizing long-term medical dependence.⁴Sickle cell anemia is a hemoglobin disorder caused by a mutation in the beta globin gene, in which normal hemoglobin A is replaced by abnormal hemoglobin S. This abnormal hemoglobin undergoes polymerization under deoxygenated conditions, resulting in rigid, sickle-shaped red blood cells.⁷ These structurally altered cells exhibit reduced deformability, leading to impaired passage through small blood vessels, microvascular occlusion, tissue hypoxia, and recurrent episodes of acute pain. The disease follows an autosomal recessive inheritance pattern, requiring both parents to be carriers for disease manifestation in offspring. It is most prevalent among populations of African, Mediterranean, and certain Asian origins.⁸
Conventional therapeutic strategies for sickle cell anemia primarily focus on symptomatic management, including pharmacological treatment, blood transfusion, and hematopoietic stem cell transplantation.⁹ Although these approaches improve survival and reduce complications, they do not address the underlying genetic abnormality.Genome editing technologies based on clustered regularly interspaced short palindromic repeats and CRISPR associated protein 9 have introduced a transformative approach by enabling direct modification at the genetic level.⁹ In sickle cell disease, these techniques are predominantly applied ex vivo to autologous hematopoietic stem cells to either correct the causative mutation or induce fetal hemoglobin expression to compensate for defective adult hemoglobin.¹⁰ Several gene editing-based therapies have progressed through clinical trials and received regulatory approval, demonstrating promising outcomes as potential functional cures for sickle cell disease.¹¹
The purpose of this review is to comprehensively analyze gene therapy approaches for sickle cell disease, with a particular focus on genome editing strategies, their mechanisms of action, therapeutic potential, clinical outcomes, and associated challenges.
Pathophysiology of Sickle Cell Disease
Sickle cell disease (SCD) is a Mendelian condition with a clear cascade of molecular and cellular events from mutation to severe systemic clinical complications.12The Pathophysiology of the disease combines hemoglobin polymerization, red blood cell deformation, vaso-occlusion, chronic hemolysis, inflammation and continued organ damage.13,14
Genetic mutation
SCD is due to a point mutation in the beta-globin (HBB) gene on chromosome 11 where a single nucleotide substitution (GAG → GTG) occurs at codon 6. This mutation replaces the glutamic acid at position 6 of the β-globin chain with valine, giving rise to hemoglobin S (HbS) that has significantly different biochemical and biophysical characteristics compared to normal adult hemoglobin (HbA). HbS is inherited in the homozygous (HbSS) state by most people with sickle cell disease, but other heterozygous states involving different β-globin mutations can also result in clinically apparent disease.6
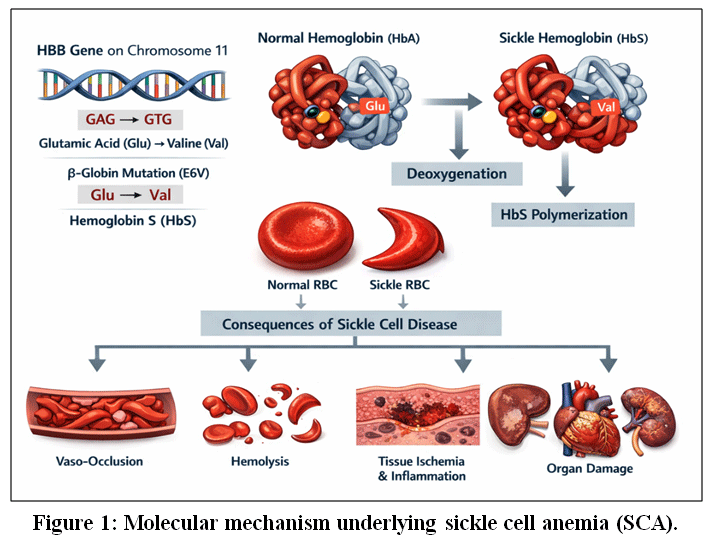 |
Figure 1: Molecular mechanism underlying sickle cell anemia (SCA).
|
Legend for Fig. 1: Sickle cell anemia results from a single point mutation in the β-globin (HBB) gene on chromosome 11, where a nucleotide substitution (GAG → GTG) leads to the replacement of glutamic acid with valine at the sixth position of the β-globin chain. This missense mutation produces abnormal hemoglobin S (HbS), which polymerizes under deoxygenated conditions. HbS polymerization causes red blood cells to adopt a rigid, sickle-shaped morphology, reducing deformability and promoting vaso-occlusion, hemolysis, chronic inflammation, tissue ischemia, and progressive multi-organ damage characteristic of sickle cell disease.6,12–14
Hemoglobin S Polymerization
In the deoxygenated state, a hydrophobic valine residue conferred by the HBB point mutation facilitates self-association of HbS molecules and results in polymerization of HbS into short, stiff rods. The polymerization of HbS is very sensitive to intracellular hemoglobin concentration; the higher the HbS, the infinitely increased formation of polymers. Crucially, fetal hemoglobin (HbF) powerfully prevents polymerization by blocking fiber extension. The HbS polymers, in turn cause damage to the red blood cell membrane, leading to cellular dehydration and a rise in intracellular viscosity while severely impairing erythrocyte deformability causing them to undergo membrane damage and become fragile.15
Sickling and Vaso-occlusion of Red Blood Cells
Polymerization of deoxygenated hemoglobin S alters red blood cells from their normal biconcave discoid shape into rigid, sickle-shaped structures. Red blood cells in affected individuals exhibit abnormal rheological properties, including increased adhesion to the vascular endothelium and reduced deformability. As a result, these cells are unable to pass efficiently through the microcirculation and tend to obstruct small blood vessels.⁶Vaso-occlusion is further promoted by enhanced adhesion of sickled red blood cells, leukocytes, and platelets to activated endothelial cells. These interactions lead to impaired blood flow, localized hypoxia, and tissue ischemia. Repeated cycles of sickling and unsickling cause progressive and irreversible damage to the red blood cell membrane, ultimately resulting in premature destruction of red blood cells and chronic hemolysis.¹⁵Collectively, these pathological processes contribute to recurrent painful vaso-occlusive episodes, which represent the primary cause of acute morbidity in sickle cell disease.⁶
Complications of Sickle Cell Disease
The later clinical manifestations of SCD are a consequence of perpetual hemolysis and episodic vaso-occlusion.
These complications include:
Hemolytic anemia: Sickled erythrocytes have a much shorter life span (10–20 days) than normal erythrocytes and cause chronic hemolytic anemia from premature destruction of the red cells.
Vaso-occlusive crises: Acute severe pain related to microvascular occlusion and hypoxic-tissue-injury.
End-Organ Damage Repeated vascular occlusion and hypoxia lead to cumulative damage to organs with high oxygen requirements including, but not limited to, the lungs (can cause acute chest syndrome), brain (stroke), kidneys (renal dysfunction), bones (e.g., avascular necrosis of femoral or humoral heads), spleen (splenic sequestration), and liver.
Endothelial dysfunction and inflammation: Released free hemoglobin, heme during hemolysis scavenges nitric oxide to cause impaired vasodilation, along with oxidative stress and inflammation by induction of the Toll-like receptor 4 (TLR-4) and inflammasome pathway.6 Greater susceptibility to infection: Repetitive splenic infarction results in functional asplenia, making patients at a much higher risk for life-threatening bacterial infections.15
Conventional Management Approaches
Conventional management modalities of sickle cell disease (SCD) are focused on decreasing the frequency and severity of clinical events, improving quality of life, and prolonging survival.13 Although these strategies have significantly advanced the treatment of cystic fibrosis over the last few decades, they are primarily supportive and fail to directly address the cause of the disease.14 Further, each of these traditional therapies has significant drawbacks and thus the requirement for more curative gene-based therapy.16,17
Hydroxyurea Therapy
Hydroxyurea is an instrumental disease-modifying agent for sickle cell disease (SCD) that functions predominantly through stimulation of fetal hemoglobin (HbF) production. High HbF levels lessen the polymerization of sickle hemoglobin, resulting in fewer blood cell sickling episodes and fewer severe vaso-occlusive crises. Clinical trials have shown that hydroxyurea treatment reduces hospitalizations, lowers the rate of acute chest syndrome and death in patients with SCD. Although it has shown benefit for many patients, the response to hydroxyurea can be highly variable and its tolerability is often limited by toxicities including myelosuppression and other hematologic toxicities, especially in subsets of patients.18
Blood Transfusion Therapy
Blood transfusion in the form of episodic and chronic transfusions is a key part of treatment for sickle cell disease by reducing the percentage of circulating sickled cells. It is the most successful in preventing and treating the most severe complications, including stroke, acute chest syndrome and severe anemia. Transfusion can be life-saving, but chronic transfusion presents an inherent risk characterized by alloimmunization, iron overload and transfusion-related infection. Therefore, chronic transfusionists need close follow-up and iron chelating therapy can ameliorate these complications.18
Hematopoietic Stem Cell Transplantation
Hematopoietic stem cell transplantation (HSCT) is the only proven curative therapy for sickle cell disease. Selected patients can be treated with allogeneic hematopoietic stem cell transplantation to eliminate disease manifestations and prevent long-term complications. But HSCT is not universally available, restricted by donor availability, the possibility of graft-versus-host disease, and transplant-related morbidity and mortality in addition to financial costs. While the introduction of new transplant technologies has resulted in improved patient outcomes, many patients remain ineligible for transplantation and long-term complications are a significant issue.17
Need for gene-based cures
The shortcomings of traditional management strategies have led to rapid developments in gene therapy and genome-editing applications in sickle cell disease.16 New therapeutic approaches, based on the gene, intend to correct or functionally replace the mutated β-globin gene.19 These methods involve gene addition with lentiviral vectors, targeted genome editing with CRISPR/Cas9 and novel gene-modification mechanisms.20,21
Gene therapy has the potential of being a definitive, one-time treatment not associated with immunological challenges inherent in allogeneic HSCT. The recent approvals of gene therapies including Casgevy and Lyfgenia highlight the promise that these technologies hold and are steps towards curative modalities for sickle cell disease (SCD) patients.22 With the development of genetic-based therapy, current research focuses on finding better delivery with lower percentages of viral nanoparticle, safety in long term, and increasing access to varied populations worldwide. Combined, these innovations mean better, easier and real one-time fixes for sickle cell disease.22
Principles of Gene Therapy
As previously described, gene therapy is defined as the modification and regulation of genetic material within a patient’s cells to prevent, cure, or mitigate an underlying genetic disorder. Instead of addressing only clinical symptoms, this approach targets the molecular basis of disease. Successful gene therapy relies on three essential components: a therapeutic payload such as a functional gene, ribonucleic acid molecule, or genome editing machinery; a delivery system or vector to transport this payload into target cells; and a treatment strategy that determines whether genetic modification is performed directly within the body (in vivo) or outside the body followed by reinfusion (ex vivo).²³
Vectors Used in Gene Therapy: Lentiviral Vectors versus Adeno-Associated Virus
Lentiviral Vectors
Lentiviral vectors function by integrating therapeutic deoxyribonucleic acid into the genome of target cells. This stable genomic integration enables long-term and sustained expression of the therapeutic gene, particularly in dividing cells such as hematopoietic stem and progenitor cells. These vectors possess a relatively large cloning capacity of approximately eight to ten kilobases, allowing incorporation of full-length genes along with their regulatory elements.
Ex vivo gene modification represents the primary clinical application of lentiviral vectors. In this approach, autologous hematopoietic stem and progenitor cells or immune cells are isolated, transduced with lentiviral vectors under good manufacturing practice conditions, evaluated for quality control, and subsequently reinfused into the patient. The risk of insertional mutagenesis associated with genomic integration has been significantly reduced through the development of self-inactivating vector designs and continuous monitoring of integration sites.²³
des-Associated Virus
Adeno-associated virus vectors represent an alternative system for gene delivery with distinct biological characteristics. Unlike lentiviral vectors, these vectors do not integrate into the host genome and instead persist primarily as episomal structures within target cells.²⁶,²⁷ This property makes them particularly suitable for non-dividing tissues such as ocular tissue, skeletal muscle, and certain regions of the central nervous system.²⁸,²⁹ Additionally, specific serotypes such as adeno-associated virus 9 exhibit tissue-specific targeting, enhancing delivery efficiency in in vivo applications.³⁰,³¹
However, adeno-associated virus vectors have a limited genetic carrying capacity of approximately 4.7 kilobases, restricting the size of therapeutic genes that can be delivered. When ex vivo manipulation is not feasible, these vectors are commonly used for in vivo gene transfer through direct tissue injection or systemic administration. Despite their advantages, systemic delivery may trigger immune responses, and pre-existing antibodies can reduce therapeutic efficacy.6,24,25
Gene Therapy Mechanisms: Gene Addition Versus Gene Replacement
Gene Addition
Gene addition involves the introduction of a functional gene copy into target cells where the endogenous gene is defective or absent. This approach does not correct the mutated gene but provides an additional source of functional protein through stable genomic integration or episomal expression.¹⁹ In sickle cell disease, gene addition strategies involve the transfer of modified beta globin genes that produce anti-sickling hemoglobin, thereby reducing hemoglobin polymerization and red blood cell sickling.²⁰ Gene addition is most effective in conditions where supplementation with a functional protein can alleviate disease pathology.¹⁹
Gene Replacement and Precision Genome Editing
Gene replacement therapies utilize genome editing technologies to directly modify the patient’s deoxyribonucleic acid. Systems based on clustered regularly interspaced short palindromic repeats and CRISPR associated protein 9, along with emerging base editors and prime editors, enable precise correction of pathogenic mutations or modulation of regulatory elements. This approach is particularly relevant for sickle cell disease, where editing the beta globin gene mutation or modifying regulatory pathways can restore normal hemoglobin function.³
These interventions are most commonly performed ex vivo in autologous hematopoietic stem and progenitor cells, allowing careful quality control before reinfusion. Genome editing techniques function by introducing targeted changes in deoxyribonucleic acid, followed by activation of cellular repair mechanisms that facilitate precise genetic modification. Additionally, editing of regulatory regions such as the erythroid-specific enhancer of B cell lymphoma eleven A can induce fetal hemoglobin expression and provide significant therapeutic benefit.⁹
Gene Therapy Approaches to Sickle Cell Anemia
Gene therapy strategies for sickle cell anemia target the disease at the molecular level by modifying autologous hematopoietic stem and progenitor cells.³² These approaches are broadly classified into two categories: gene addition using anti-sickling globin genes and genome modification strategies aimed at increasing fetal hemoglobin production.³
Gene Addition: Therapeutic Beta-Like Globin Gene Delivery
Gene addition approaches involve the delivery of modified beta globin genes that encode anti-sickling hemoglobin. One such variant, hemoglobin A T87Q, is engineered to function similarly to normal hemoglobin while preventing polymerization under hypoxic conditions. In this method, hematopoietic stem and progenitor cells are isolated from the patient and transduced ex vivo with lentiviral vectors to achieve stable integration of the therapeutic gene. Following reinfusion, these modified cells continuously produce red blood cells expressing therapeutic hemoglobin, providing long-term clinical benefit.
The LentiGlobin platform represents one of the most advanced examples of this approach. Clinical studies have demonstrated sustained expression of therapeutic hemoglobin, reduction in vaso-occlusive episodes, and decreased dependence on blood transfusions.³
Induction of Fetal Hemoglobin through Modulation of B Cell Lymphoma Eleven A
An alternative strategy involves the induction of fetal hemoglobin, which effectively inhibits polymerization of hemoglobin S.³³ Elevated levels of fetal hemoglobin reduce intracellular hemoglobin S concentration and decrease red blood cell sickling, as observed in individuals with hereditary persistence of fetal hemoglobin.³⁴
B cell lymphoma eleven A acts as a key transcriptional repressor of gamma globin gene expression after birth.³⁴ Targeted suppression of this regulator in erythroid cells results in increased fetal hemoglobin production without affecting its function in other tissues.³⁵
Two primary approaches are used:
Ribonucleic Acid-Based Silencing
Ribonucleic acid interference techniques utilize short hairpin ribonucleic acid constructs to reduce expression of B cell lymphoma eleven A at the post-transcriptional level. This results in sustained fetal hemoglobin production and significant clinical improvement.³⁶
Genome Editing of Regulatory Elements
Genome editing techniques disrupt regulatory regions controlling B cell lymphoma eleven A expression, leading to reactivation of fetal hemoglobin. This selective approach increases fetal hemoglobin levels specifically in red blood cells while preserving normal function in other tissues.³⁵
This strategy is exemplified by exagamglogene autotemcel, in which autologous hematopoietic stem and progenitor cells are edited ex vivo to disrupt the erythroid-specific enhancer of B cell lymphoma eleven A. Following reinfusion, these cells produce red blood cells with high levels of fetal hemoglobin, resulting in significant clinical improvement and near elimination of vaso-occlusive episodes.³
Crispr Based Therapies for Sickle Cell Anemia
Clustered regularly interspaced short palindromic repeats and CRISPR associated protein 9 based genome editing has emerged as a highly precise and efficient therapeutic strategy for sickle cell anemia by enabling targeted modification of disease-associated genetic loci.³² Unlike traditional gene addition methods, this approach allows direct manipulation of endogenous genes and regulatory elements, leading to sustained and physiologically regulated therapeutic effects.³⁵
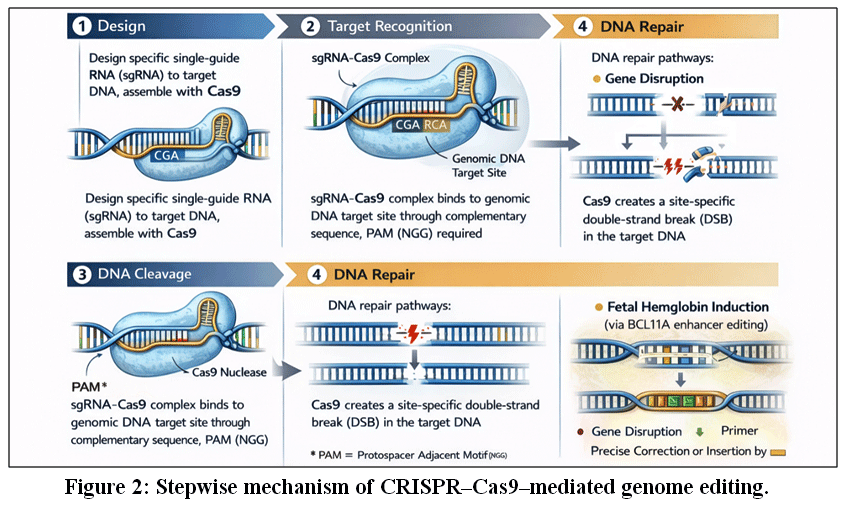 |
Figure 2: Stepwise mechanism of CRISPR–Cas9–mediated genome editing.
|
Legend for Fig. 2 -The CRISPR–Cas9 genome-editing process involves four coordinated steps. (1) Design: A single-guide RNA (sgRNA) is engineered to be complementary to a specific target DNA sequence and assembled with the Cas9 endonuclease. (2) Target recognition: The sgRNA–Cas9 complex binds to the genomic target site through sequence complementarity, guided by the presence of a protospacer-adjacent motif (PAM). (3) DNA cleavage: Cas9 introduces a site-specific double-strand break (DSB) at the target locus. (4) DNA repair: Endogenous cellular DNA repair pathways resolve the break via non-homologous end joining (NHEJ), resulting in gene disruption, or homology-directed repair (HDR), enabling precise gene correction or insertion. This programmable system forms the molecular basis of CRISPR-based gene therapies for sickle cell disease, including targeted disruption of regulatory elements such as the BCL11A enhancer to induce fetal hemoglobin expression.2,3,9,10,37,38
CTX001 (Exagamglogene Autotemcel)
CTX001, also known as exagamglogene autotemcel, is an investigational autologous gene-edited hematopoietic stem cell therapy developed by Vertex Pharmaceuticals and CRISPR Therapeutics for the treatment of severe sickle cell disease and transfusion-dependent beta thalassemia. In this approach, CD34 positive hematopoietic stem and progenitor cells are isolated from patients and edited ex vivo to inactivate the erythroid-specific enhancer of the B cell lymphoma eleven A gene. This targeted disruption results in selective repression of B cell lymphoma eleven A in erythroid cells, thereby reactivating fetal hemoglobin production and effectively preventing hemoglobin S polymerization.³
In a first-in-human clinical trial, electroporation of CD34 positive cells with CRISPR associated protein 9 ribonucleoprotein complexes targeting the B cell lymphoma eleven A enhancer achieved approximately eighty percent allelic modification at the target site. Following reinfusion and one year of follow-up, patients demonstrated sustained expression of fetal hemoglobin in both bone marrow and peripheral blood, with widespread distribution across erythrocytes. Clinically, patients achieved transfusion independence or complete elimination of vaso-occlusive crises. Importantly, detailed molecular analysis did not detect significant off-target genome editing events using the available assays.³
Subsequent studies involving larger patient populations and extended follow-up periods have confirmed sustained fetal hemoglobin induction, clinical efficacy, and an acceptable safety profile. These findings have supported the advancement of this therapy into pivotal trials and regulatory approvals in multiple regions, establishing it as a first-in-class genome editing-based therapy for sickle cell disease.¹⁹
Advantages of CRISPR Based Therapy for Sickle Cell Disease
CRISPR based therapies offer several advantages over conventional gene addition approaches. First, disruption of the B cell lymphoma eleven A enhancer restores the endogenous fetal hemoglobin program, providing a natural protective mechanism against red blood cell sickling. Fetal hemoglobin is uniformly distributed across erythrocytes, ensuring consistent and predictable therapeutic effects.³
Second, these therapies have the potential to provide a one-time curative treatment. Ex vivo edited long-term repopulating hematopoietic stem cells continuously generate healthy red blood cells, enabling sustained therapeutic benefit throughout the patient’s lifetime.³
Third, genome editing avoids the introduction of exogenous genetic material and instead mimics naturally occurring benign genetic conditions such as hereditary persistence of fetal hemoglobin. This reduces the risk of insertional mutagenesis associated with integrating viral vectors.³
Risks and Technical Challenges
Despite their significant potential, CRISPR based therapies are associated with several technical and clinical challenges. Off-target genome editing remains a major concern, as unintended double-strand breaks may occur at genomic sites with partial sequence similarity. These events can lead to insertions, deletions, large chromosomal rearrangements, or translocations. Although improvements in guide ribonucleic acid design, high-fidelity nucleases, and chemical modifications have reduced off-target activity, complete elimination of risk has not yet been achieved.³⁹
Another major challenge is the efficient delivery of genome editing components. In sickle cell disease, ex vivo electroporation of CRISPR associated protein 9 ribonucleoprotein complexes into hematopoietic stem and progenitor cells is preferred, as it allows transient exposure and extensive quality control prior to reinfusion. In contrast, in vivo delivery remains technically challenging due to issues such as immune responses, poor targeting efficiency, and systemic toxicity.⁴⁰,⁴¹
Conditioning regimens required for engraftment, such as myeloablative chemotherapy, are associated with significant toxicity and may result in long-term adverse effects including infertility and secondary malignancies. Additionally, immune responses against genome editing components may occur, although this risk is reduced with ex vivo delivery strategies.⁴¹
Risk Mitigation Strategies
Multiple strategies have been developed to enhance the safety of genome editing therapies. These include the use of CRISPR associated protein 9 ribonucleoprotein complexes and high-fidelity nucleases to minimize exposure and reduce off-target activity. Comprehensive off-target analysis is performed using computational prediction tools and genome-wide screening techniques, along with single-cell and chromosomal analyses to detect large-scale genomic alterations prior to reinfusion.³⁷
Furthermore, ongoing clinical studies are exploring modified or reduced-intensity conditioning regimens to decrease transplantation-related toxicity while maintaining effective engraftment.
Challenges and Ethical Considerations
Despite the promising therapeutic potential of CRISPR based gene editing for sickle cell disease, several technical, clinical, economic, and ethical challenges must be addressed before widespread clinical implementation can be achieved.⁴²
Delivery Challenges
Efficient and safe delivery of genome editing components remains a critical challenge. Currently, ex vivo editing of autologous hematopoietic stem and progenitor cells represents the standard clinical approach. In this process, patient-derived stem cells are collected, edited using genome editing tools, thoroughly evaluated for safety and efficacy, and then reinfused following conditioning therapy. This approach allows detailed assessment of editing efficiency, off-target effects, and genomic stability before administration.⁴³
However, ex vivo procedures require specialized infrastructure, strict adherence to good manufacturing practice standards, complex logistics, and conditioning regimens, limiting scalability and global accessibility. In contrast, in vivo genome editing has the potential to simplify treatment and expand access, although significant challenges remain, including targeted delivery, immune compatibility, and control over gene editing activity.⁴⁴,⁴⁵
Off-Target Effects and Genome Integrity
CRISPR based genome editing introduces targeted double-strand breaks in deoxyribonucleic acid; however, unintended cleavage at non-target sites remains a significant safety concern. Such off-target effects may lead to insertions, deletions, or chromosomal rearrangements, particularly in long-lived hematopoietic stem cells, potentially resulting in clonal expansion and malignancy.⁴⁶⁻⁴⁸
To address these risks, rigorous safety measures are incorporated into clinical protocols, including optimized guide ribonucleic acid design, genome-wide off-target screening methods such as GUIDE sequencing, CIRCLE sequencing, and SITE sequencing, and the use of high-fidelity nucleases. Additionally, deep sequencing and cytogenetic analyses are performed prior to reinfusion. Despite these precautions, long-term monitoring of treated patients remains essential.⁴⁹,⁵⁰
Cost and Accessibility
Gene and cell therapies are the most costly therapeutics that exist today. The personalized nature of autologous interventions, together with GMP-compliant manufacturing, conditioning regimens, hospitalization long-term follow-up, can translate into costs often exceeding hundreds of thousands of dollars per single patient. This is a herculean task for sickle cell disease, particularly when one realizes that the population burden of this disease falls disproportionately on low- and middle-income countries and isolated communities.51
In addition to cost, lack of infrastructure—insufficient treatment centers, trained staff, and reliable cold-chain systems also prevents access. Several policy responses, including outcomes-based payment systems, annuity-like payments, and public–private partnerships, are considered to increase affordability. Yet in the absence of parallel innovation in manufacturing scale, streamlined treatment regimens, and global health investment, these therapies will paradoxically contribute to widening disparities in health. Therefore, equitable access has to be postulated as an intrinsic part of the scientific production.51
Ethical Considerations and Germline Editing
Genome editing also provokes urgent ethical questions, in particular those about the distinction between somatic and germline modifications. Somatic gene editing, which does not affect the next generation of the treated person, is widely considered to be ethically permissible when performed under stringent safety and regulatory oversight. Editing the germ line—making changes to embryos, eggs, or sperm that would be passed from parent to child—presents vastly more complicated ethical problems.53
In the aftermath of a widely criticized germline editing experiment that took place in China in 2018, international scientific and regulatory organizations have called for strict limits or even a ban on clinical germline editing pending the satisfactory resolution of safety, regulatory, and societal consensus issues, include uncertainty about long-term effects, the absence of consent from prospective generations, justice and equity issues, and misuse of technology for non-therapeutic enhancement or for eugenic purposes.53
Understandably, CRISPR-based approaches to treating sickle cell disease today — of which CTX001/exa-cel for SCD is just one example — are entirely somatic and do not entail germline modulation. Nonetheless, continuing ethics monitoring, open public debate, and strong regulation are crucial as genome-editing methods develop.42
Future Prospects
The rapid development of genome editing technologies has made gene therapy an achievable and potentially curative treatment option for sickle cell disease (SCD).32Current active innovation aims to advance precision, safety, access, and global scalability to facilitate the longevity of clinical success.
More Retrainable Genome Surgeons
Both base editors and prime editors, novel genome-editing systems developed in the next wave of genome editing, constitute a significant step forward compared to traditional CRISPR/Cas9 technology.54,55 These technologies facilitate targeted alteration of DNA without creation of double-strand breaks (DSBs), with the result that the risk of large deletions, chromosomal rearrangement, and genomic instability is greatly reduced.57
Base editors induce direct base alterations, including C→T and A→G changes, that are particularly well suited for the correction of disease-related point mutations.54 Prime editors are even more flexible and can achieve precise insertions, deletions, and all combinations of the four possible base substitutions with a minimal level of off-targets.55 Both approaches have shown robust efficacy in preclinical models.52,54,55efforts are ongoing to enhance delivery, efficiency, and specificity in HSPCs.57
If issues relating to delivery efficiency and off-target base-editing activity can be addressed, these systems may allow direct correction of the single nucleotide substitution underlying sickle cell disease or accurate targeting of regulatory elements such as BCL11A, providing safer and more targeted therapeutic options.57
Key takeaway: Base and prime editing are equipotent to diminish DSB-related hazards, yet broadening the possibilities regarding the limited scope of exact genomic tinkering, hence warrant further investigation as serious contenders towards the clinical armamentarium.
In Vivo Editing of Hematopoietic Stem Cells
An ultimate long-term objective in sickle cell therapies is the creation of facilities for in vivo hematopoietic stem and progenitor cell (HSPC) editing that would obviate the requirement to harvest, manipulate, and infuse ex vivo stem cells.44In vivo editing has the potential to dramatically simplify treatment regimens while decreasing cost and increasing access worldwide.51
Nevertheless, major obstacles still need to be overcome, such as optimal targeting of bone marrow stem cell niches, prevention of systemic toxicities and immune escape, and efficient engraftment by the gene‐edited long-term repopulating HSC.40 New developments in targeted nanoparticles and transient viral and non-viral delivery indicate that in vivo genome editing is biologically possible.57 However, a series of preclinical validation and safety profiling is necessary before these approaches can be brought to the clinic.51
Systems-Level Responses: Production, Financing, and Global Equity
Scientific advances alone are not enough to deliver broad impact, unless there are parallel developments in health-care systems. Manufacturing innovations (e.g., automation, decentralized or regional GMP facilities, and rationalization of production workflows) will be necessary to scale gene-editing therapies to minimize expense and complexity.51
Equally vital are the development of sustainable financing models that relate payment to long-term clinical outcome, including outcomes-based reimbursement models, annuity-style payment mechanisms, and global public–private partnerships.51 International cooperation and funding for infrastructure, workforce training, and harmonization of regulations will be required to enable gene-editing therapy to reach the populations most affected by SCD, particularly in LMICs.51,56
Conclusion
Clustered regularly interspaced short palindromic repeats and CRISPR associated protein 9 based genome editing, along with next-generation editing technologies, has ushered in a transformative era in the management of sickle cell disease by offering clinically viable, potentially curative single-treatment strategies. By enabling reactivation of fetal hemoglobin through targeted inactivation of B cell lymphoma eleven A, or precise correction of the pathogenic mutation within the beta globin gene locus, these approaches move beyond symptomatic management toward true disease modification. Early clinical outcomes, particularly with genome editing therapies such as exagamglogene autotemcel, demonstrate sustained reduction in vaso-occlusive episodes and meaningful long-term improvement in patient outcomes.
The rationale for this review lies in the need to critically evaluate these rapidly advancing therapeutic strategies, integrate current mechanistic and clinical evidence, and identify the translational challenges that may influence their broader clinical adoption. While gene editing technologies hold unprecedented promise, their implementation is constrained by several key challenges, including delivery limitations, immunogenicity, unintended genomic alterations, conditioning-related toxicity, high treatment costs, and disparities in global accessibility.
Based on the evidence presented, it can be hypothesized that continued advancements in precision genome editing, safer delivery platforms, and optimized conditioning strategies will enable gene therapy to evolve into a standardized curative approach for sickle cell disease. However, achieving this goal will require not only scientific innovation but also robust ethical governance, equitable healthcare policies, and scalable manufacturing systems.
In conclusion, the transition of gene editing from experimental therapy to routine clinical practice represents a realistic and attainable objective, provided that ongoing scientific, regulatory, and societal challenges are addressed in a coordinated and sustainable manner.
Acknowledgement
The authors would like to acknowledge the academic support and constructive suggestions provided by colleagues and mentors during the preparation of this review.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable.
Author Contributions
- Vaishnavi Vinod Dere: Conceptualization, literature review, data collection, writing – original draft preparation, and manuscript organization.
- Sushant Satappa Patil: Literature survey, data compilation, reference management, and writing review & editing.
- Ajay Yeshawant Kale: Scientific guidance, methodology development, critical revision of the manuscript, and supervision.
- Kishor Vasant Otari: Project administration, overall supervision, final review, and approval of the manuscript for publication.
References
- High KA, Roncarolo MG. Gene therapy. N Engl J Med. 2019;381(5):455–464. doi:10.1056/NEJMra1706910
CrossRef - Doudna JA, Charpentier E. Genome editing with CRISPR-Cas9. Science. 2014;346(6213):1258096. doi:10.1126/science.1258096
CrossRef - Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. N Engl J Med. 2021;384(3):252–260. doi:10.1056/NEJMoa2031054
CrossRef - Singh A, Irfan H, Fatima E, Nazir Z, Verma A, Akilimali A. FDA approval of CRISPR therapy for sickle cell disease. Ann Med Surg. 2024;86:4555–4559. doi:10.1016/j.amsu.2024.01.012
CrossRef - S. Food and Drug Administration. FDA approves gene therapies for sickle cell disease. 2023.
- Sundd P, Gladwin MT, Novelli EM. Pathophysiology of sickle cell disease. Annu Rev Pathol. 2019;14:263–292. doi:10.1146/annurev-pathmechdis-012418-012838
CrossRef - Rees DC, Williams TN, Gladwin MT. Sickle cell disease. Lancet. 2010;376(9757):2018–2031. doi:10.1016/S0140-6736(10)61029-X
CrossRef - Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. doi:10.1038/nrdp.2018.10
CrossRef - Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;376(16):1561–1573. doi:10.1056/NEJMra1510865
CrossRef - Park SH, Lee CM, Deshmukh H, Bao G. CRISPR genome editing for sickle cell disease. Blood. 2016;128(22):4703
CrossRef - Chaudhary A, Kumari B, Choudhury M, et al. CRISPR in sickle cell disease. Int J Sci Res Publ. 2021;11(1):670–676
CrossRef - Mohammed AT. CRISPR gene therapy for sickle cell disease. J Hematol Clin Res. 2025;9(1):11–14
CrossRef - Taher AT, Weatherall DJ, Cappellini MD. Hemoglobinopathies. Lancet. 2025
- Orkin SH, Bauer DE. Genetic therapy for sickle cell disease. Annu Rev Med. 2019;70:257–271. doi:10.1146/annurev-med-041817-044229
CrossRef - Bhatia M, Sheth S. Stem cell transplantation in sickle cell disease. J Blood Med. 2015;6:229–238. doi:10.2147/JBM.S60564
- Weaver JM, Ashcroft MT, Telfer P. Management of sickle cell disease. Br J Haematol. 2024;204(2):267–280
- Cavazzana M, Corsia A, Brusson M, et al. Gene therapy approaches. Annu Rev Pharmacol Toxicol. 2025;65:397–413
CrossRef - Ribeil JA, Hacein-Bey-Abina S, Payen E, et al. Gene therapy in sickle cell disease. N Engl J Med. 2017;376(9):848–855. doi:10.1056/NEJMoa1609677
CrossRef - Naldini L. Gene therapy advances. Nature. 2015;526(7573):351–360. doi:10.1038/nature15818
CrossRef - Six E, Cavazzana M. Stem cell gene therapy. Mol Ther. 2023;31(1):3–15
- Naso MF, Tomkowicz B, Perry WL, et al. Adeno-associated virus vectors in gene therapy. BioDrugs. 2017;31(4):317–334
CrossRef - Wang D, Tai PWL, Gao G. Adeno-associated virus delivery systems. Nat Rev Drug Discov. 2019;18(5):358–378
CrossRef - Daya S, Berns KI. AAV gene therapy vectors. Clin Microbiol Rev. 2008;21(4):583–593
CrossRef - Colella P, Ronzitti G, Mingozzi F. AAV clinical issues. Mol Ther Methods Clin Dev. 2017;8:87–104. doi:10.1016/j.omtm.2017.03.007
CrossRef - Mendell JR, Al-Zaidy S, Shell R, et al. Gene therapy clinical trial. N Engl J Med. 2017;377(18):1713–1722. doi:10.1056/NEJMoa1706198
CrossRef - Hudry E, Vandenberghe LH. Therapeutic gene transfer to the CNS. Neuron. 2019;102(2):263–275
CrossRef - Zincarelli C, Soltys S, Rengo G, et al. Analysis of AAV serotypes. Mol Ther. 2008;16(6):1073–1080
CrossRef - Bevan AK, Duque S, Foust KD, et al. Systemic gene delivery. Mol Ther. 2011;19(11):1971–1980
CrossRef - Sankaran VG, Orkin SH. Hemoglobin switching. Cold Spring Harb Perspect Med. 2013;3(1):a011643
CrossRef - Sankaran VG, Xu J, Ragoczy T, et al. BCL11A regulation. Nature. 2009;460:1093–1097
CrossRef - Canver MC, Smith EC, Sher F, et al. CRISPR enhancer editing. Nature. 2015;527:192–197
CrossRef - Brendel C, Guda S, Renella R, et al. BCL11A targeting. J Clin Invest. 2016;126:3868–3878
CrossRef - Mengstie MA, Azezew MT, Dejenie TA. Off-target CRISPR effects. Biologics. 2024;18:21–28
CrossRef - Cai L, Bai H, Mahairaki V, et al. HBB mutation correction. Stem Cells Transl Med. 2018;7:87–97
CrossRef - Guo C, Ma X, Gao F, et al. CRISPR off-target analysis. Front Bioeng Biotechnol. 2023;11:1143157
CrossRef - Li L, Hu S, Chen X. CRISPR delivery systems. Biomaterials. 2019;207:1–12
CrossRef - Luther DC, Lee YW, Nagaraj H, et al. CRISPR delivery strategies. Expert Opin Drug Deliv. 2019;16:160–172
- Rubeis G, Steger F. Genome editing ethics. Asian Bioeth Rev. 2018;10:133–141
CrossRef - Dever DP, Bak RO, Reinisch A, et al. β-globin gene editing. Nature. 2016;539:384–389
CrossRef - Behr M, Zhou J, Xu B, et al. In vivo CRISPR delivery. Acta Pharm Sin B. 2021;11:2150–2171
CrossRef - Yin H, Kauffman KJ, Anderson DG. Delivery technologies for genome editing. Nat Rev Drug Discov. 2024;23(2):98–117
- Tsai SQ, Joung JK. Genome editing specificity and safety. Nat Rev Genet. 2023;24(4):215–232
- Kosicki M, Tomberg K, Bradley A. DNA repair outcomes. Nat Biotechnol. 2023;41(6):765–774
CrossRef - Cornu TI, Mussolino C, Cathomen T. Clinical translation of genome editing. Nat Med. 2024;30(1):12–21
- Vakulskas CA, Dever DP, Rettig GR, et al. High-fidelity genome editing. Nat Med. 2023;29(8):1456–1464
- Cornetta K, Bonamino M, Mahlangu J, et al. Global access to gene therapy. Mol Ther. 2024;32(6):2100–2112
- Cyranoski D. Ethical concerns in human genome editing. Nature. 2023;620(7972):20–22
CrossRef - National Academies of Sciences. Human genome editing guidelines update. Washington DC; 2024
- Komor AC, Levy JM, Liu DR. Base editing developments. Nature. 2023;615(7950):420–430
- Anzalone AV, Koblan LW, Liu DR. Prime editing advances. Nature. 2023;618(7965):676–685
- Gillmore JD, Maitland ML, Lebwohl D, et al. Clinical genome editing therapies. N Engl J Med. 2024;390(5):410–420
- Caso F, Davies B. Base and prime editing applications. J Histochem Cytochem. 2023;71(9):623–638
- Musunuru K, Chadwick AC, Mizoguchi T, et al. In vivo base editing therapy. Nature. 2023;615(7952):430–435
- Newby GA, Liu DR. Next-generation genome editing tools. Cell. 2024;187(3):500–515
- Porteus MH. Clinical applications of genome editing. Nat Rev Genet. 2024;25(2):85–102
- Ledford H. CRISPR therapies moving to clinic. Nature. 2025;630(8001):15–18
- Zhang F, Cong L. Advances in CRISPR technologies. Science. 2026;371(6535):eabc1234
Abbreviations
SCD – Sickle Cell Disease
SCA – Sickle Cell Anemia
RBC – Red Blood Cell
Hb – Hemoglobin
HbS – Hemoglobin S
HbA – Hemoglobin A
HbF – Fetal Hemoglobin
HBB – β-globin gene
CRISPR – Clustered Regularly Interspaced Short Palindromic Repeats
Cas9 – CRISPR-associated protein 9
sgRNA – Single Guide RNA
DSB – Double-Strand Break
NHEJ – Non-Homologous End Joining
HDR – Homology-Directed Repair
HSPCs – Hematopoietic Stem and Progenitor Cells
HSCT – Hematopoietic Stem Cell Transplantation
AAV – Adeno-Associated Virus
LV – Lentiviral Vector
GMP – Good Manufacturing Practice
CTX001 / exa-cel – Exagamglogene Autotemcel
VOC – Vaso-Occlusive Crisis
TLR-4 – Toll-Like Receptor 4
Accepted on: 16-04-2026
Second Review by: Dr. Kirti Dubli
Final Approval by: Dr. Eugene A. Silow








![]()
![]()
