
Redox Imbalance and Oxidative Stress in Diabetes Mellitus: Mechanistic Insights into Complications and Antioxidant-Based Therapeutic Perspectives
 , Nabiha Zameer2, Pargi Shruthi2and Nyalata Archita2
, Nabiha Zameer2, Pargi Shruthi2and Nyalata Archita2 1Department of Pharmacology, Joginpally B.R. Pharmacy College, Yenkapally, Moinabad, Hyderabad, India.
2Department of Pharmacy, Joginpally B.R. Pharmacy College, Yenkapally, Moinabad, Hyderabad, India.
Corresponding Author E-mail: mahi.unaj@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3484
Download this article as:
![]()
Diabetes mellitus (DM) is a metabolic disorder marked by chronic hyperglycemia and characterized by micro- and macrovascular complications. Oxidative stress, a state of redox imbalance, is an important etiological factor in the pathophysiology of insulin resistance, β-cell glucotoxicity, inflammation, and cardiovascular complications of diabetes, resulting from the accumulation of reactive oxygen and nitrogen species (ROS/RNS). Chronically elevated glucose levels lead to the generation of reactive oxygen species (ROS) through glucose auto-oxidation, advanced glycation end-product formation, and activation of the polyol pathway and protein kinase C (PKC) signaling. Insulin signaling is also compromised by oxidative stress through inhibition of insulin signaling pathways, suppression of GLUT4-dependent glucose uptake, and stimulation of stress kinases and inflammatory pathways. Pancreatic β-cells, owing to their low antioxidant defense, are particularly vulnerable to oxidative damage, ultimately leading to their functional exhaustion. The purpose of this review is to discuss the contribution of exogenous and endogenous antioxidants in maintaining redox homeostasis, protecting β-cells, improving insulin sensitivity, modulating inflammation, and preventing vascular complications. Emerging strategies such as Nrf2-mediated antioxidant signaling, nano-antioxidants, and targeted antioxidant delivery systems show promising potential for improving glycemic control and reducing diabetes-related complications through redox-sensitive therapeutic approaches.
KEYWORDS:Antioxidant signaling; Antioxidant defense; Hyperglycemia; Redox homeostasis
Introduction
Diabetes mellitus (DM) is a chronic metabolic condition caused by either insufficient insulin synthesis by the pancreas or impaired insulin responsiveness in the body1. β cells, which are located in the pancreatic islets of Langerhans, produce insulin, a hormone that is essential for regulating blood sugar levels in humans2. Long-term complications such as neuropathy, nephropathy, retinopathy, and atherosclerosis can arise from prolonged hyperglycemia in diabetics.3-5
Types of Diabetes Mellitus
The American Diabetes Association (ADA) classifies diabetes mellitus into four specific types, notably gestational diabetes mellitus (GDM), type 1 diabetes mellitus (T1DM), and type 2 diabetes mellitus (T2DM). 6 T1DM is characterized by the autoimmune destruction of pancreatic β-cells, resulting in an absolute deficiency of insulin, necessitating lifelong insulin therapy.7, 8 More than 90% of diabetes cases are type 2 diabetes mellitus (T2DM), which is characterized by insulin resistance with progressive β-cell dysfunction and is directly linked to obesity, ageing, and chronic metabolic inflammation. Pharmacological therapies and lifestyle changes can be utilised to treat T2DM. 9 Gestational insulin resistance leads to gestational diabetes mellitus (GDM), a pregnancy-related glucose intolerance, which increases a woman’s risk for acquiring type 2 diabetes and cardiovascular disease.10,11
Other Types of Diabetes
Mature-Onset Diabetes of the Young (MODY): A monogenic form caused by hereditary abnormalities in insulin secretion. Proper genetic identification is emphasized in ADA guidelines. 12
Neonatal Diabetes: Resulting from mutations leading to severe insulin deficiency in infants under 6–12 months, affecting growth and neurological development. 13
Type 3c Diabetes: Arises from pancreatic exocrine disease (e.g., chronic pancreatitis, pancreatic cancer) causing insulin deficiency and resistance. 14
Latent Autoimmune Diabetes in Adults (LADA): Slowly developing autoimmune diabetes in adults, diagnosed via C-peptide levels and autoantibodies. 15
Cystic Fibrosis-Related Diabetes: Features both insulin resistance and deficiency due to CF pathophysiology.16
Steroid-Induced Diabetes: Hyperglycemia due to glucocorticoid therapy causing β-cell dysfunction and insulin resistance.17
Insulin Signaling Pathway
Insulin is a pancreatic β-cell hormone secreted in response to postprandial glucose elevation and plays a major role in maintaining glucose homeostasis. It promotes glucose uptake in insulin-sensitive tissues by stimulating GLUT4 translocation and regulates key metabolic processes, including glycogen synthesis, lipid metabolism, and suppression of gluconeogenesis. Glucose-stimulated insulin secretion is mediated by ATP-dependent membrane depolarization, calcium influx, and insulin exocytosis. 18,19
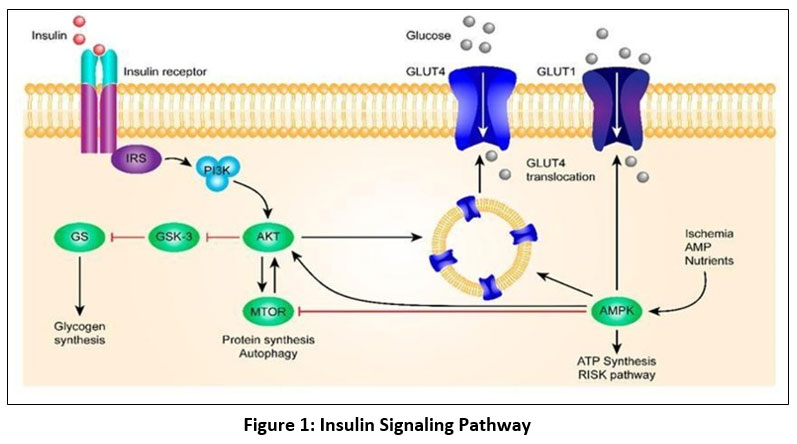 |
Figure 1: Insulin Signaling Pathway
|
Glycogen production, inhibition of lipolysis, stimulation of lipogenesis, suppression of gluconeogenesis, and increased glucose absorption are only a few of the metabolic activities that insulin controls. A signaling complex is formed beneath the membrane when insulin interacts to its cell-surface insulin receptor (IR). The IR is mostly found in muscle, liver, and adipose tissues. It is a heterotetramer made up of two external α-subunits and two intracellular β-subunits. Receptor tyrosine kinase activity is triggered by insulin binding, which also results in IR dimerization and ATP binding.20 The Ras–MAPK route (cell growth/differentiation) and the PI3K–AKT pathway (metabolic consequences) are the two main signaling pathways that are started when the active receptor autophosphorylates and phosphorylates insulin receptor substrates (IRS).
By converting PIP2 to PIP3, PI3K activates PDK1/PDK2, which in turn activates AKT/PKB and atypical PKCs. By phosphorylating AS160, activated AKT enhances the absorption of glucose in muscle and adipose tissue via increasing GLUT4 translocation to the plasma membrane. In order to encourage the synthesis of glycogen in the liver and skeletal muscle, AKT also suppresses GSK3.21,22
Insulin Resistance and the Central Role of Oxidative Stress in Diabetes
Insulin resistance is a main pathogenic feature of type 2 diabetes mellitus and precedes β-cell dysfunction and chronic hyperglycaemia. Under normal conditions, glucose homeostasis is tightly regulated; however, defects in insulin signaling disrupt this balance. Insulin resistance arises from alterations in insulin receptor (IR) and insulin receptor substrate (IRS) function, leading to impaired PI3K/AKT signaling and reduced GLUT4-mediated glucose uptake. These defects are driven by abnormal serine/threonine phosphorylation of IRS proteins, increased activity of protein tyrosine phosphatases such as PTP1B, mitochondrial dysfunction, endoplasmic reticulum stress, inflammation, and trigger of stress-responsive kinases including JNK, IKK, and PKC 23-25.
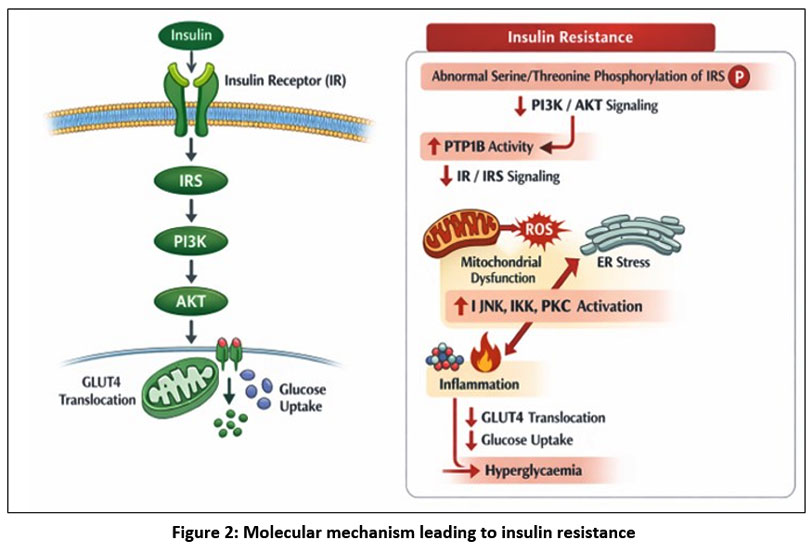 |
Figure 2: Molecular mechanism leading to insulin resistance
|
Mechanisms of Oxidative Stress and Insulin Resistance
Oxidative stress plays a central role in the disruption of insulin signaling associated with type 2 diabetes mellitus.26 Excessive production of reactive oxygen and nitrogen species (ROS/RNS) leads to abnormal serine/threonine phosphorylation of insulin receptor substrate (IRS) proteins and subsequent impaired downstream phosphatidylinositol 3-kinase (PI3K)–Akt signaling and substantially decreased GLUT4-mediated glucose uptake in insulin-sensitive tissues.27,28
Increased ROS levels potentiate stress-responsive kinases such as c-Jun N-terminal kinase (JNK), inhibitor of κB kinase β (IKKβ), protein kinase C (PKC), and p38 mitogen-activated protein kinase (MAPK).29 Activation of these kinases interferes with insulin receptor signaling and enhances nuclear factor-κB (NF-κB)–mediated inflammatory responses, contributing to chronic low-grade inflammation and insulin resistance.30,31
In adipose tissue, oxidative stress alters adipokine secretion. It reduces adiponectin levels, a key adipokine that enhances insulin sensitivity, while increasing the production of pro-inflammatory cytokines such as TNF-α and IL-6.32 Consequently, oxidative stress contributes to systemic insulin resistance, mitochondrial dysfunction, and metabolic dysregulation, thereby promoting the progression of type 2 diabetes mellitus.33
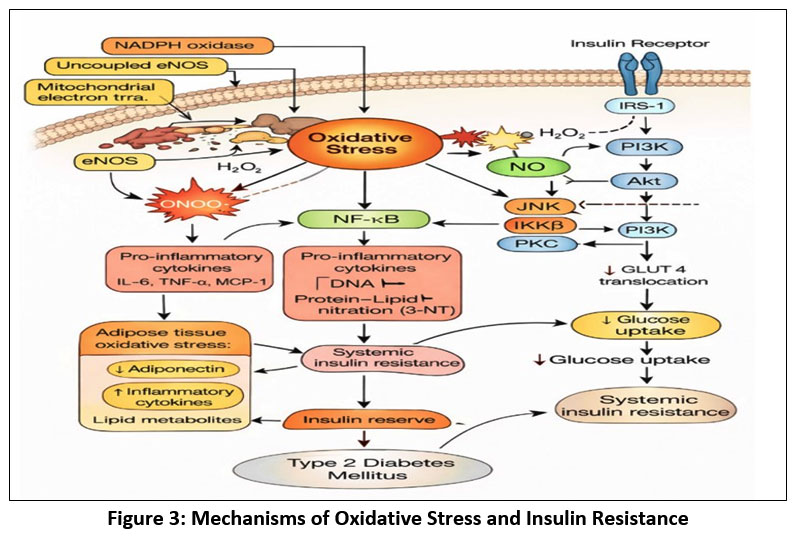 |
Figure 3: Mechanisms of Oxidative Stress and Insulin Resistance
|
Insulin signaling disruption brought on by oxidative stress is a hallmark of type 2 diabetes. Reactive oxygen and nitrogen species generated by mitochondrial malfunction, NADPH oxidase, and uncoupled eNOS activate NF-κB and stress kinases (JNK, IKKβ, PKC). These mechanisms limit GLUT4 translocation and glucose uptake, disrupt IRS-1/PI3K/Akt signaling, produce pro-inflammatory cytokines and adipokines, and ultimately result in systemic insulin resistance and type 2 diabetes mellitus.
β-Cell Dysfunction and Oxidative Stress
Due to their low antioxidant capacity, pancreatic β-cells are particularly susceptible to oxidative damage. Persistent oxidative stress impairs glucose-stimulated insulin secretion, damages mitochondrial DNA, activates inflammatory signaling pathways, and promotes β-cell apoptosis, ultimately resulting in β-cell exhaustion and failure. Persistent ROS exposure impairs glucose-stimulated insulin secretion, damages mitochondrial DNA, and triggers apoptotic pathways, leading to β-cell exhaustion and decreased β-cell mass. Oxidative stress can also disturb transcription factors critical for β-cell function like PDX-1 and MafA, further declining insulin biosynthesis and release.
Influence on Inflammation and Endothelial Function
Insulin activates Akt, phosphorylates insulin receptor substrate-1 (IRS-1), PI3-kinase, and PIP2, and promotes the insulin receptor signaling pathway, resulting in the translocation of glucose transporter 4 (GLUT4) to the plasma membrane. Inflammation of the liver and adipose tissue brought on by obesity stimulates the generation of ROS, proinflammatory cytokines, and macrophage infiltration. Increased ROS causes insulin resistance and hyperglycemia by blocking the insulin receptor signaling pathway.
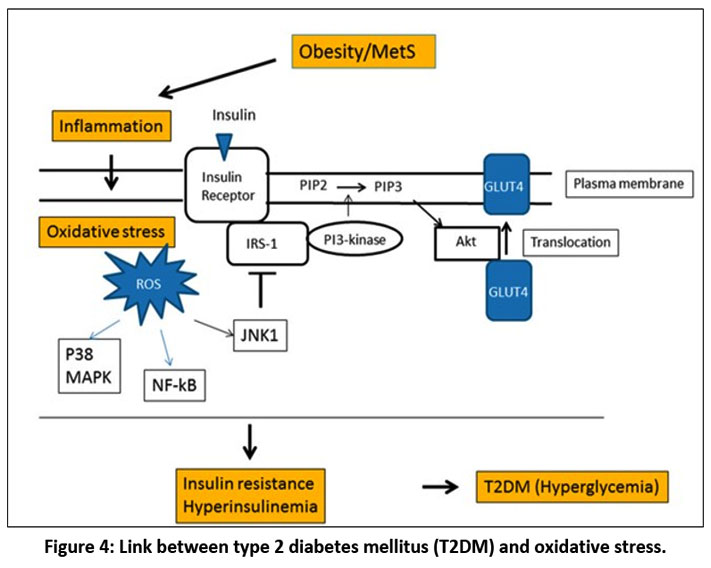 |
Figure 4: Link between type 2 diabetes mellitus (T2DM) and oxidative stress.
|
Antioxidant Defense and Oxidative Stress in Diabetes
Antioxidants, both endogenous and exogenous, are prerequisites for maintaining redox homeostasis by neutralizing reactive oxygen species (ROS) and modulating oxidative signaling pathways. Under healthy circumstances, oxidative damage is restricted by enzymatic antioxidants such as glutathione peroxidase, catalase, and superoxide dismutase as well as non-enzymatic defenses like glutathione.; however, in diabetes, these systems become overwhelmed, reflected by a reduced GSH/GSSG ratio, a hallmark of oxidative stress and insulin resistance. While physiological ROS participate in normal metabolic signaling, chronic hyperglycaemia promotes excessive ROS generation via glucose autoxidation, AGE formation, protein kinase C activation, and mitochondrial dysfunction, with pancreatic β-cells being particularly vulnerable due to low antioxidant capacity. Antioxidants counteract these effects by scavenging ROS, suppressing oxidative pathways, preserving β-cell function, promoting insulin sensitivity, and slowing the progression of diabetic complications.
Mechanisms by Which Antioxidants Prevent and Ameliorate Diabetes
Protection of pancreatic β-cells by antioxidants
Pancreatic β-cells are particularly at risk to oxidative stress due to their low endogenous antioxidant capacity. Chronic hyperglycaemia and elevated free fatty acids increase mitochondrial ROS production, resulting in oxidative damage that disrupts` insulin biosynthesis and secretion. Antioxidants protect β-cells mainly by scavenging reactive oxygen species and restoring redox balance, thus preserving mitochondrial integrity and ATP generation essential for glucose-stimulated insulin secretion.
A key protective mechanism necessitates activation of the Nrf2–ARE pathway, which strengthens transcription of cytoprotective and antioxidant genes, including HO-1, NQO1, and glutathione-related enzymes. In addition, antioxidants suppress oxidative stress–induced β-cell apoptosis and inflammation by inhibiting stress-activated kinases like JNK and p38 MAPK, reducing endoplasmic reticulum stress, and diminishing NF-κB–mediated inflammatory signaling. Collectively, these effects preserve β-cell mass, preserve critical transcription factors such as PDX-1 and MafA, and sustain insulin gene expression under diabetic conditions.
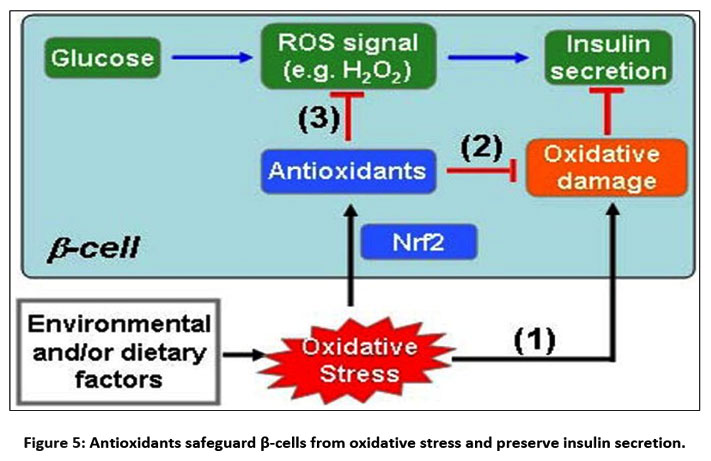 |
Figure 5: Antioxidants safeguard β-cells from oxidative stress and preserve insulin secretion.
|
(1) Oxidative stress brought on by environmental and/or dietary factors causes oxidative damage, which directly reduces beta-cell function; (2) Oxidative stress triggers the Nrf2-mediated antioxidant response, which shields cells from oxidative damage; and (3) Enhanced antioxidant capacity by Nrf2 activation adversely affects glucose-derived ROS signaling, which is a component of glucose-stimulated insulin secretion GSIS.
Improvement of Insulin Sensitivity
Insulin resistance, the hallmark of type 2 diabetes mellitus, is closely associated with oxidative stress–induced impairment of insulin signaling in skeletal muscle, adipose tissue, and liver. Excess reactive oxygen species produced during chronic hyperglycaemia and lipotoxicity stimulate inhibitory serine phosphorylation of insulin receptor substrates, resulting in impaired PI3K–Akt signaling, decreased GLUT4 translocation, and diminished glucose uptake.
Antioxidants mitigate these effects by scavenging ROS and preserving the functional integrity of insulin receptors and downstream signaling components.In addition, antioxidants suppress ROS-activated stress kinases, including JNK, IKKβ, and p38 MAPK, which disrupt insulin signaling and promote inflammation. By inhibiting NF-κB–mediated cytokine production, particularly TNF-α and IL-6 in adipose tissue, antioxidants attenuate chronic low-grade inflammation and improve systemic insulin sensitivity.
Activation of AMP-activated protein kinase (AMPK), a key cellular energy sensor, that functions independently of insulin signaling, is another significant way that antioxidants increase insulin sensitivity. Dietary antioxidants such as polyphenols, flavonoids, and α-lipoic acid activate AMPK by improving cellular redox status and mitochondrial efficiency. AMPK activation enhances GLUT4 translocation, increases fatty acid oxidation, suppresses hepatic gluconeogenesis, and reduces lipid accumulation in insulin-responsive tissues, thereby alleviating lipid-induced inhibition of insulin signaling.
Antioxidants also improve mitochondrial function by limiting mitochondrial ROS overproduction and restoring oxidative phosphorylation efficiency. Improved mitochondrial bioenergetics reduce oxidative damage to mitochondrial DNA and proteins, prevent mitochondrial-driven insulin resistance, and support normal glucose and lipid metabolism.
Compounds such as α-lipoic acid and polyphenols further promote mitochondrial redox balance and biogenesis through regeneration of endogenous antioxidants and modulation of transcriptional regulators such as PGC-1α, collectively improving whole-body insulin sensitivity.
Dietary and endogenous antioxidants improve insulin sensitivity by limiting oxidative damage to insulin receptors and signaling proteins, enhancing GLUT4 expression and translocation, activating AMP-activated protein kinase (AMPK), and improving mitochondrial function and cellular energy metabolism. Among these, polyphenols, flavonoids, and α-lipoic acid have demonstrated particular efficacy in restoring insulin responsiveness in experimental model.
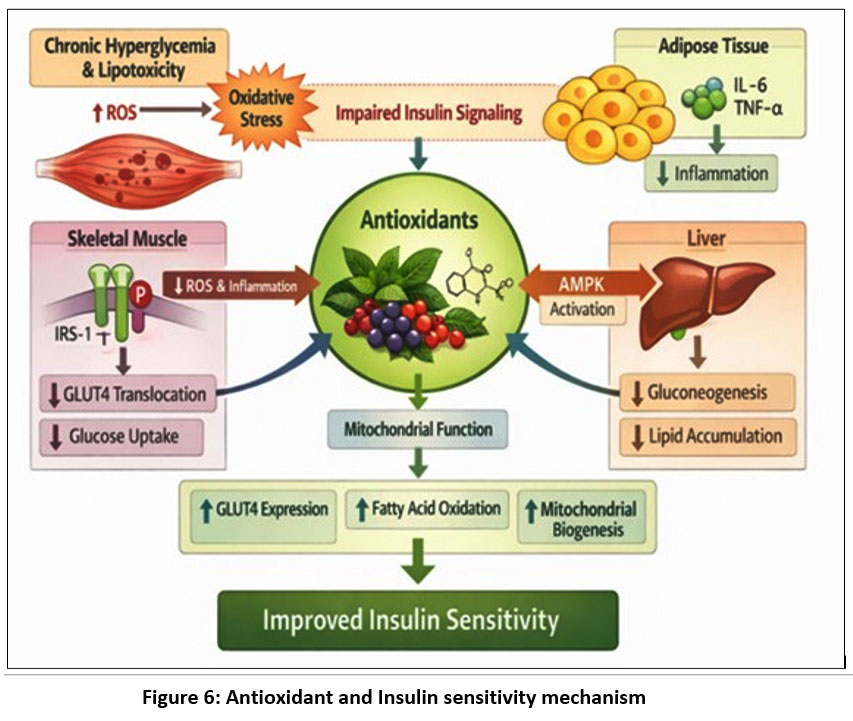 |
Figure 6: Antioxidant and Insulin sensitivity mechanism
|
Chronic hyperglycemia and lipotoxicity elevate reactive oxygen species (ROS), leading to oxidative stress and impaired insulin signaling. Antioxidants reduce ROS and inflammation in skeletal muscle, enhance AMPK activation in the liver, and suppress pro-inflammatory cytokines in adipose tissue. These actions improve mitochondrial function, increase GLUT4 expression, promote fatty acid oxidation, and support mitochondrial biogenesis.
Regulation of Inflammation and Endothelial Function
Chronic low-grade inflammation in diabetes is closely linked to oxidative stress, as reactive oxygen species activate pro-inflammatory transcription factors such as NF-κB, increasing cytokine and adhesion molecule expression and promoting endothelial dysfunction and vascular complications. Antioxidants impair these effects by inhibiting ROS-dependent inflammatory signaling and restoring nitric oxide bioavailability, thereby improving endothelial function, reducing lipid peroxidation, and normalizing vascular reactivity in diabetic conditions.
Types of Antioxidants and Their Roles in Diabetes
Endogenous Antioxidants
Enzymatic antioxidants, including SOD, CAT, GPx, and glutathione reductase, constitute the primary defense against oxidative stress. In diabetes, altered activity of these enzymes results in redox imbalance and increased oxidative damage.
Non-enzymatic antioxidants such as glutathione, uric acid, and albumin directly scavenge ROS and maintain intracellular redox status. A reduced GSH/GSSG ratio is a consistent marker of oxidative stress and insulin resistance in diabetic patients.
Dietary and Natural Antioxidants
Dietary antioxidants play a complementary role in reinforcing endogenous defenses. These include:
Vitamins: Vitamin C regenerates other antioxidants and reduces glycation; vitamin E protects membrane lipids from peroxidation.
Polyphenols and flavonoids: Found in fruits, vegetables, herbs, and spices, these compounds modulate oxidative stress, inflammation, and glucose metabolism.
Carotenoids and terpenoids: Contribute to antioxidant and anti-inflammatory activity.
Epidemiological studies consistently associate antioxidant-rich diets with reduced incidence of diabetes and its complications.
Functional Foods and Plant-Derived Antioxidants
Herbs, spices, medicinal plants, and agricultural by-products are rich sources of bioactive antioxidants. Compounds such as curcumin, catechins, resveratrol, and oleuropein activate antioxidant enzymes and suppress oxidative damage. With additional metabolic and anti-inflammatory advantages, these natural antioxidants provide a safer and more sustainable substitute for synthetic antioxidants.
Recent Advancements in Antioxidant-Based Diabetes Therapy
Nrf2-Mediated Antioxidant Signaling
A major advancement in antioxidant research is the identification of the Nrf2-ARE pathway as a master regulator of cellular antioxidant defenses. Activation of Nrf2 enhances transcription of cytoprotective genes such as HO-1, NQO1, and GCLC, improving cellular resistance to oxidative stress. Natural compounds like resveratrol, sulforaphane, and epigallocatechin gallate have been shown to activate Nrf2 and protect diabetic tissues.
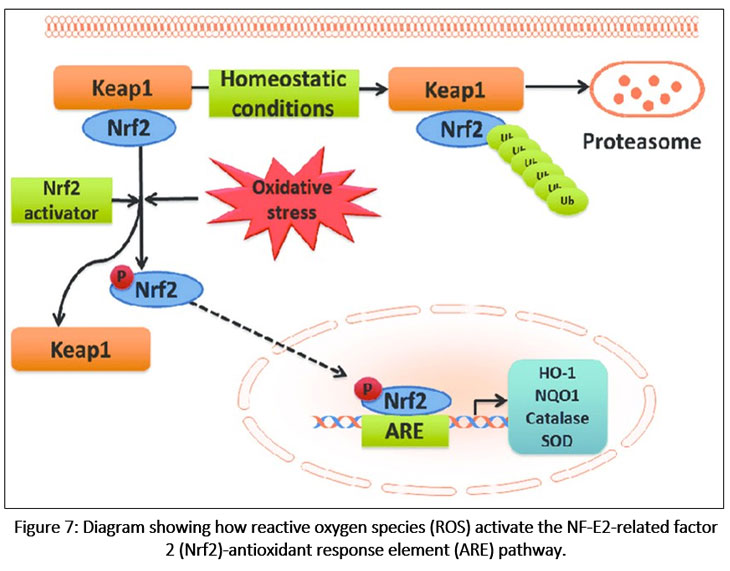 |
Figure 7: Diagram showing how reactive oxygen species (ROS) activate the NF-E2-related factor 2 (Nrf2)-antioxidant response element (ARE) pathway.
|
During normal condition, Nrf2 is constitutively linked to the Keap1 protein in the cytoplasm. By encouraging Nrf2 ubiquitination and subsequent proteasomal pathway breakdown, Keap1 suppresses the Nrf2 signaling pathway. Nrf2-Keap1 complex breakup, Nrf2 phosphorylation, and nuclear translocation are all brought on by mild oxidative stress and Nrf2 activators. By attaching to the ARE in the target genes’ promoter regions, Nrf2 stimulates the transcriptional activation of detoxification enzymes and antioxidants (heme oxygenase-1 [HO-1], NAD(P) H:quinone oxidoreductase 1 [NQO1], catalase, and superoxide dismutase [SOD]).
Nanotechnology and Targeted Antioxidant Delivery
One limitation of antioxidant therapy is poor bioavailability and tissue targeting. Recent approaches involve nanoparticle-based delivery systems, liposomes, and polymeric carriers to enhance antioxidant stability, permeability, and targeted delivery to pancreatic β-cells, nerves, and retinal tissue. These strategies represent promising advancements in improving clinical efficacy.
Nanotechnology has also provided access through delivery of antioxidants and also successful strategies targeting important regulatory elements/ signallers to support pancreatic β-cell regeneration. By delivering a polymeric nano-delivery containing an anti-GSK-3β silencing agent, Wnt/β-catenin signaling is enhanced to promote β-cell proliferation and survival. The inhibition of GSK3β results in the lack of degradation of β-catenin, allowing translocation to the nucleus and transcriptional activation of genes responsible for β-cell growth and insulin production. Therefore, nanotechnology appears to play an important role in redox modulation as well as β-cell regeneration therapy.
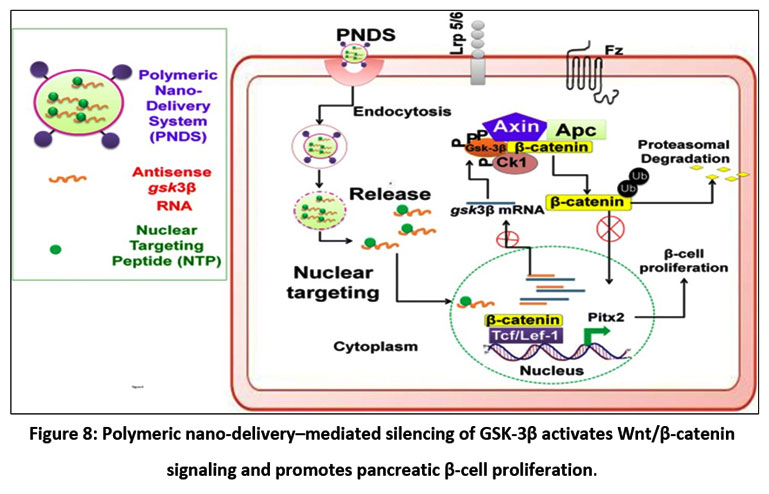 |
Figure 8: Polymeric nano-delivery–mediated silencing of GSK-3β activates Wnt/β-catenin signaling and promotes pancreatic β-cell proliferation.
|
The polymeric nano-delivery system (PNDS) carrying antisense GSK-3β RNA and a nuclear-targeting peptide (NTP) enters cells via endocytosis. After endosomal release, the RNA is directed to the nucleus by NTP, suppressing GSK-3β mRNA. This inhibition prevents β-catenin phosphorylation and degradation, allowing its nuclear translocation to activate TCF/LEF-1 target genes, including Pitx2, thereby promoting β-cell proliferation. PNDS-mediated GSK-3β silencing shows potential for β-cell regeneration.
Combination Therapies
Emerging evidence suggests that antioxidants are most effective when used in combination with standard antidiabetic drugs or other antioxidants. Synergistic effects have been observed with combinations like vitamin C, vitamin E, and α-lipoic acid, targeting multiple oxidative pathways simultaneously while minimizing adverse effects.
The Antioxidant Paradox and Challenges in Clinical Translation
Despite strong experimental evidence demonstrating the role of oxidative stress in diabetes pathogenesis, many clinical studies using traditional antioxidant supplements have shown no clear benefits and, in some cases, adverse outcomes.34 This is supported by major clinical trials such as HOPE, 35 SELECT,36 and ACCORD.37 Several studies have reported that β-carotene and vitamin E supplementation do not reduce cardiovascular events in individuals with diabetes and may increase the risk of adverse outcomes. This phenomenon is referred to as the ‘antioxidant paradox’ 38,39 and demonstrates that ROS can be both positive and negative within the human body.
Physiological levels of ROS40 are required for proper cellular signaling, including insulin signaling and glucose-stimulated insulin secretion. Using high doses of exogenous antioxidant products can impede the normal functioning of physiological cellular activities and thereby reverse any potential for their therapeutic abilities.
A distinction must be made between strategies that enhance endogenous antioxidant defenses and exogenous antioxidant supplementation. In particular, methods to stimulate the activity of the body’s natural (intrinsic) antioxidant system, through Nrf2-mediated gene expression for antioxidants,41,42,43 have a greater likelihood of success compared to using direct scavengers of reactive oxygen species (ROS).44,45 Antioxidants may also have significantly greater benefits when used early in the process leading to insulin resistance than when used late in the progression of diabetes when vascular complications are already present.46,47
The findings of these studies emphasize the need for specific, mechanism-based approaches to antioxidant therapy in diabetes management, rather than non-specific antioxidant supplementation.
Conclusion
Antioxidants have a significant role in preventing diabetes onset and progression by protecting pancreatic β-cells, improving insulin sensitivity, reducing inflammation, and preserving vascular function. While experimental and early clinical studies highlight significant therapeutic potential, inconsistent results in large clinical trials underscore the need for optimized dosing strategies, improved delivery systems, and long-term human studies. Future research should focus on targeted antioxidant therapies, synergistic combinations, and personalized approaches to effectively integrate antioxidants into diabetes management.
Acknowledgement
The authors would like to thank Joginpally B.R. Pharmacy College for providing institutional support and facilities.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable
Author Contributions
Dr.B.Maheshwari– Supervised the entire work and wrote the final draft.
Nabiha Zameer– Completed formatting.
P.Shruthi– Completed Literature review.
N.Archita– Data editing.
References
- World Health Organization. Global report on diabetes. World Health Organization; 2016.
- American Diabetes Association. Standards of care in diabetes—2023. Diabetes Care. 2023;46(Suppl 1):S1-S291. doi:10.2337/dc23-SINT
CrossRef - International Diabetes Federation. IDF Diabetes Atlas. 10th ed. International Diabetes Federation; 2021.
- DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am. 2004;88(4):787-835. doi:10.1016/j.mcna.2004.04.013
CrossRef - Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329(14):977-986. doi:10.1056/NEJM199309303291401
CrossRef - American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2014;37(Suppl 1):S81-S90. doi:10.2337/dc14-S081
CrossRef - Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. 2014;383(9911):69-82. doi:10.1016/S0140-6736(13)60591-7
CrossRef - Van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91(1):79-118. doi:10.1152/physrev.00003.2010
CrossRef - Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet. 2014;383(9922):1068-1083. doi:10.1016/S0140-6736(13)62154-6
CrossRef - Metzger BE, Gabbe SG, Persson B, et al. International Association of Diabetes and Pregnancy Study Groups recommendations. Diabetes Care. 2010;33(3):676-682. doi:10.2337/dc09-1848
CrossRef - Buchanan TA, Xiang AH. Gestational diabetes mellitus. J Clin Invest. 2005;115(3):485-491. doi:10.1172/JCI24531
CrossRef - Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic β-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4(4):200-213. doi:10.1038/ncpendmet0778
CrossRef - Edghill EL, Flanagan SE, Patch AM, et al. Insulin mutation screening in diabetes. Diabetes. 2008;57(4):1034-1042. doi:10.2337/db07-1405
CrossRef - Hardt PD, Ewald N. Exocrine pancreatic insufficiency in diabetes mellitus. Lancet Diabetes Endocrinol. 2013;1(3):226-235. doi:10.1016/S2213-8587(13)70046-X
- Tuomi T, Groop LC, Zimmet PZ, et al. Antibodies to glutamic acid decarboxylase reveal latent autoimmune diabetes in adults. Diabetes. 1999;48(1):12-19. doi:10.2337/diabetes.48.1.12
- Moran A, Pyzdrowski KL, Weinreb J, et al. Insulin resistance in cystic fibrosis–related diabetes. Diabetes Care. 2010;33(12):2697-2708. doi:10.2337/dc10-1769
CrossRef - Clore JN, Thurby-Hay L. Glucocorticoid-induced hyperglycemia. Endocr Pract. 2009;15(5):469-474. doi:10.4158/EP08331.RAR
CrossRef - Ashcroft FM, Rorsman P. Diabetes mellitus and the β cell: the last ten years. Cell. 2012;148(6):1160-1171. doi:10.1016/j.cell.2012.02.010
CrossRef - Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycemia of diabetes mellitus. Endocr Rev. 1998;19(4):491-503. doi:10.1210/edrv.19.4.0336
CrossRef - Saltiel AR, Kahn CR. Insulin signaling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799-806. doi:10.1038/414799a
CrossRef - Whiteman EL, Cho H, Birnbaum MJ. Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab. 2002;13(10):444-451. doi:10.1016/S1043-2760(02)00662-1
CrossRef - DeFronzo RA. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis. Diabetes Care. 2010;33(7):166-172. doi:10.2337/dc10-s070
CrossRef - Petersen KF, Shulman GI. Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes. Am J Cardiol. 2002;90(5A):11G-18G. doi:10.1016/S0002-9149(02)02968-8
CrossRef - Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852-871. doi:10.1016/j.cell.2012.02.017
CrossRef - Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793-1801. doi:10.1172/JCI29069
CrossRef - Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways. Endocr Rev. 2002;23(5):599-622. doi:10.1210/er.2001-0039
CrossRef - Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440(7086):944-948. doi:10.1038/nature04634
CrossRef - Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860-867. doi:10.1038/nature05485
CrossRef - Ceriello A, Motz E. Is oxidative stress the pathogenic mechanism underlying insulin resistance and β-cell dysfunction? Arterioscler Thromb Vasc Biol. 2004;24(5):816-823. doi:10.1161/01.ATV.0000122852.22604.78
CrossRef - Tilg H, Moschen AR. Inflammatory mechanisms in the regulation of insulin resistance. Nat Rev Immunol. 2006;6(10):772-783. doi:10.1038/nri1931
CrossRef - Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med. 2006;119(5 Suppl 1):S10-S16. doi:10.1016/j.amjmed.2006.01.009
CrossRef - Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks diabetic damage. Nature. 2000;404(6779):787-790. doi:10.1038/35008121
CrossRef - Hoehn KL, Salmon AB, Hohnen-Behrens C, et al. Insulin resistance is a cellular antioxidant defense mechanism. Proc Natl Acad Sci U S A. 2009;106(42):17787-17792. doi:10.1073/pnas.0902380106
CrossRef - Bast A, Haenen GRMM. Ten misconceptions about antioxidants. Curr Top Med Chem. 2013;13(12):1377-1390. doi:10.2174/15680266113136660156
CrossRef - Evans JL, Goldfine ID. Alpha-lipoic acid: a multifunctional antioxidant. Diabetes. 2000;49(7):1057-1060. doi:10.2337/diabetes.49.7.1057
- Zhang WJ, Frei B. Alpha-lipoic acid inhibits oxidative stress–induced NF-κB activation. FASEB J. 2001;15(13):2423-2432. doi:10.1096/fj.01-0266com
CrossRef - Ceriello A. Oxidative stress and glycemic regulation. Metabolism. 2000;49(2 Suppl 1):27-29. doi:10.1016/S0026-0495(00)80083-0
CrossRef - Radak Z, Chung HY, Goto S. Systemic adaptation to oxidative challenge. Free Radic Biol Med. 2008;44(2):153-159. doi:10.1016/j.freeradbiomed.2007.01.029
CrossRef - Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27(3):813-823. doi:10.2337/diacare.27.3.813
CrossRef - Devaraj S, Jialal I. Alpha-tocopherol supplementation and endothelial function. Diabetes Care. 2000;23(3):392-398. doi:10.2337/diacare.23.3.392
- Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients. N Engl J Med. 2000;342(3):154-160. doi:10.1056/NEJM200001203420302
CrossRef - Klein EA, Thompson IM Jr, Tangen CM, et al. Vitamin E and the risk of prostate cancer. JAMA. 2011;306(14):1549-1556. doi:10.1001/jama.2011.1437
CrossRef - Action to Control Cardiovascular Risk in Diabetes Study Group. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358(24):2545-2559. doi:10.1056/NEJMoa0802743
CrossRef - Bast A, Haenen GRMM. The antioxidant paradox. Trends Pharmacol Sci. 2013;34(8):430-436. doi:10.1016/j.tips.2013.05.007
CrossRef - Ristow M, Schmeisser K. Mitohormesis: promoting health and lifespan. Dose Response. 2014;12(2):288-341. doi:10.2203/dose-response.13-035.Ristow
CrossRef - Bellezza I, Giambanco I, Minelli A, Donato R. Nrf2–Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta Mol Cell Res. 2018;1865(5):721-733. doi:10.1016/j.bbamcr.2018.02.001
CrossRef - Forman HJ, Zhang H. Targeting oxidative stress in disease. Nat Rev Drug Discov. 2021;20(9):689-709. doi:10.1038/s41573-021-00233-1
CrossRef
Accepted on: 20-02-2026
Second Review by: Dr. Saeed Kewedar
Final Approval by: Dr. Eugene A. Silow








![]()
![]()
