
Pancreatic Lipase Inhibitors: Mechanistic Foundations, Structural Insights, and Emerging Medicinal Chemistry Strategies
 and Vipul Manusinh Vaghela2
and Vipul Manusinh Vaghela2
1Research scholar, Department of Pharmacy, Gujarat Technological University, Nr. Vishwakarma Government Engineering College, Gujarat, India
2Department of Pharmaceutical Chemistry, A. R. college of pharmacy and G. H. Patel institute of pharmacy, Gujarat, India.
Corresponding Author E-mail: nbp9171@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3478
Download this article as:
![]()
The main dietary triglyceride hydrolyzing catalyst, and a long time pharmacological parameter of decreased dietary caloric intake, is pancreatic lipase (PL). Recent progress in structural biology, high resolution crystalography and computational models has given a new understanding of the catalytic triad of PL and interfacial activation, lid dynamics and stabilization of colipase dependent. These mechanistic underpinnings have facilitated more rational search of the varied classes of inhibitors including covalent β-lactones to reversible natural products (flavonoids, aurones, chalcones) and contemporary synthetic scaffolds like thiazolidinedione, triazole, and multi-target hybrid chemotypes. The mechanism by which the inhibitors interact with the hydrophobic acyl-binding tunnel, oxyanion hole, and aromatic platform around Ser152 is now understood using quantitative structure-activity correlations, molecular docking, molecular dynamics simulations, and pharmacophore models. The new approaches to medicinal chemistry, such as allosteric inhibition of lid movement, partial inhibition to enhance the safety, the investigation of non-2-lactone electrophiles, and AI-assisted scaffold discovery provide avenues to effective, yet safer inhibitors. The enzymatic mechanism, structural biology, SAR trends, and computational methods have been incorporated in this review to present a single framework in designing next-generation pancreatic lipase inhibitors.
KEYWORDS:Allosteric inhibition; Interfacial activation; Molecular docking; Molecular dynamics; Pancreatic lipase (PL); Pharmacophore modeling; QSAR; Scaffold hopping; Structure-based drug design
Introduction
Pancreatic lipase (PL) is the principal enzyme responsible for hydrolyzing dietary triglycerides into absorbable free fatty acids and monoglycerides.1 Being the rate-limiting step of lipid assimilation, PL is a promising pharmacological target in reducing the caloric uptake and controlling obesity. However, the theoretical simplicity of the approach to inhibition of fat-digestion has not been easily converted into the safe and effective pharmacotherapy. Orlistat, the sole clinically approved inhibitor is constrained by potency to tolerability trade-offs, gastro intolerability and insignificant weight loss in the long term.2
Clinical evidence reveals that orlistat achieves modest weight reduction of approximately 2.9 kg beyond placebo over 12 months, with only 37-54% of patients maintaining adherence due to gastrointestinal adverse events including steatorrhea, fecal urgency, and fat-soluble vitamin malabsorption.2 The disconnect between biochemical potency (IC₅₀ 0.092 μM) and clinical outcomes highlights a fundamental challenge: complete lipase inhibition triggers compensatory mechanisms including increased ghrelin secretion, altered satiety signaling, and behavioral adaptation that diminish long-term efficacy.2,3 This potency-to-tolerability paradox necessitates a paradigm shift from maximal enzyme blockade toward controlled, partial inhibition strategies that preserve physiological lipid digestion while reducing caloric absorption.
Recent advances in structural biology, computational chemistry, natural-product exploration, and medicinal chemistry have revitalized interest in next-generation lipase modulators.3 A deeper understanding of interfacial activation, lid-domain dynamics, the colipase interaction surface, and substrate selectivity has uncovered new allosteric and partially inhibitory opportunities.1,4 In parallel, modern high-throughput screening, scaffold hopping, structure-guided design, and AI-assisted chemistry have expanded the chemical space5 of inhibitors far beyond β-lactones.6
This review synthesizes fundamental mechanistic insight into PL activation and catalysis, summarizes key natural and synthetic inhibitor classes, and highlights emerging medicinal-chemistry strategies — including allosteric modulation, multi-target hybrid scaffolds, and non–β-lactone electrophiles — that may overcome the limitations of classical inhibition.
Enzymatic Mechanism and Structural Biology
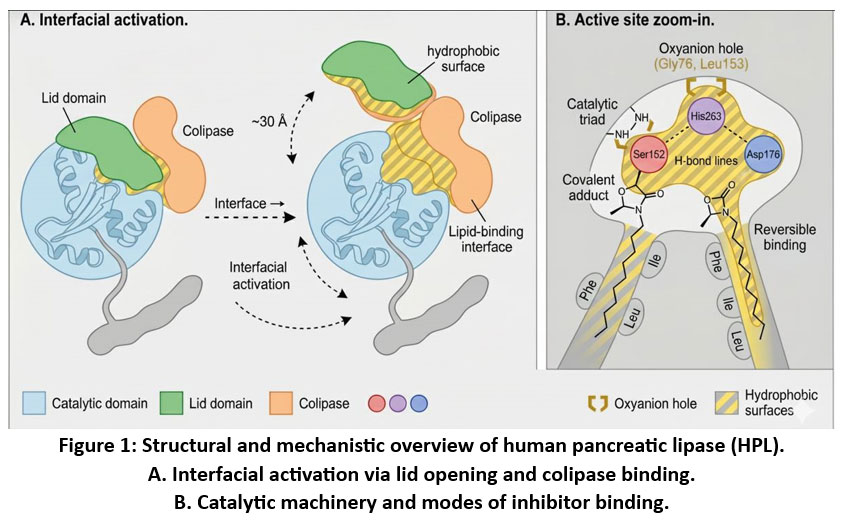 |
Figure 1: Structural and mechanistic overview of human pancreatic lipase (hPL). A. Interfacial activation via lid opening and colipase binding. B. Catalytic machinery and modes of inhibitor binding.
|
Interfacial Activation
A defining feature of pancreatic lipase is interfacial activation, the dramatic increase in catalytic activity observed when the enzyme encounters a lipid–water interface. In aqueous solution, the active site is shielded by a mobile lid domain, which adopts a closed conformation. Upon interaction with lipid droplets, conformational rearrangements shift the lid into an open state, exposing the catalytic machinery and forming a hydrophobic plateau optimized for substrate binding.7
It is activated by the action of colipase, which is a small cofactor that anchors PL to the mixed micelles in the presence of inhibitory bile salts. The interaction between the lid, colipase, and the lipid interface is critical to the effective turnover of the substrate as well as being a good target of interest when designing allosteric or partial inhibitors which seek to moderate activity rather than to block it.
Catalytic Triad and Mechanistic Chemistry
Like other serine hydrolases, PL employs a Ser–His–Asp catalytic triad (Ser152–His263–Asp176).8 Catalysis proceeds through:9,10
Nucleophilic attack by Ser152 on the ester bond of the triglyceride, forming a tetrahedral intermediate stabilized by an oxyanion hole.
Acyl-enzyme formation after collapse of the intermediate and release of the first product.
Deacylation, where water (activated by His263) cleaves the acyl–enzyme intermediate to regenerate Ser152.
PL exhibits strong regiospecificity, preferentially hydrolyzing ester bonds at the sn-1 and sn-3 positions, yielding 2-monoglycerides as dominant products. This selectivity is governed by steric constraints within the active site and by subtle lid-domain positioning that guides substrate orientation.11
Colipase Interaction and Structural Stabilization
In the intestinal lumen, bile salts form mixed micelles that can displace lipase from the lipid surface. Colipase overcomes this by binding tightly to PL, stabilizing the open lid conformation and anchoring the enzyme to micelles. The PL–colipase complex provides a broader hydrophobic surface for substrate entry. Structurally, co-crystalized complexes (e.g., PDB 1LPB and 1LPS) show how colipase contacts the C-terminal domain and helps maintain the catalytic configuration under physiological conditions.12,13
Crystal Structures and Conformational States
Multiple high-resolution structures have revealed the conformational flexibility of PL:14
1LPB: PL–colipase complex, lid closed4
1LPS: PL–colipase with a phosphonate inhibitor, lid open15
1ETH: PL bound to a substrate analogue16
1N8S: Lipase-colipase-bile salt micelle complex1
Together, these structures provide an invaluable foundation for rational inhibitor design, illuminating both the catalytic pocket and distal allosteric regions that may be exploited for partial inhibition
Classes of Pancreatic Lipase Inhibitors
Pancreatic lipase inhibition has been explored across multiple chemical spaces, reflecting the enzyme’s complex interplay of hydrophobic substrate binding, interfacial activation, and unique catalytic architecture.8
Effective inhibitors must reconcile two competing biochemical principles:3
High affinity for a deeply hydrophobic acyl-binding tunnel, and
Selective engagement with the catalytic machinery of Ser152–His263–Asp176 without inducing systemic exposure.
These dual constraints give rise to several distinct inhibitor classes, each shaped by specific structural, mechanistic, and physicochemical requirements.
β-Lactones — Covalent, Mechanistically Precise, but Carrying a Potency–Tolerability Paradox
β-Lactones represent the clearest chemical mimicry of the transition state for ester hydrolysis, explaining their unparalleled potency.17 Their strained four-membered ring positions the carbonyl exactly where the catalytic Ser152 nucleophile attacks, allowing rapid formation of a tetrahedral intermediate mimic that collapses into a stable covalent acyl-enzyme.18
Concrete mechanistic reasons for their potency
The electrophilic β-lactone carbonyl is perfectly aligned with the oxyanion hole (Phe77, Leu153).17
The long aliphatic side chain mimics a fatty acyl tail, optimally filling the lipophilic tunnel.
Side-chain stereochemistry determines whether the carbonyl oxygen can “sit” in the oxyanion hole in a transition-state–like geometry.19
Potency range
Most β-lactones show inhibition in the low nanomolar to sub-micromolar region.19
Concrete limitation
Their irreversible covalent nature and hydrophobicity lead to:2
Local gastrointestinal (GI) accumulation
Broad suppression of fat digestion
Tolerability issues (steatorrhea, oily spotting, dose-limiting symptoms)
This is why medicinal chemistry efforts increasingly target reversible or partially reversible alternatives.
Natural Product–Derived Inhibitors — Potent, Polyfunctional Chemotypes with Drug-Likeness Challenges
Natural products offer structural diversity not easily achievable synthetically. Their molecular frameworks often provide precisely positioned aromatic rings and hydrogen-bonding groups, enabling them to compete with orlistat in binding efficiency despite being reversible.
Flavonoids & Flavan-3-ols (Concrete SAR advantages)
Flavones and flavan-3-ols demonstrate inhibition by non-covalent stabilization of the lipase active site,20 mainly by
π–π stacking with Phe77 and Tyr11421
Hydrogen bonding to backbone NH groups forming the oxyanion hole.20
Hydrophobic insertion of phenyl rings into the acyl-binding tunnel.21
Potency range
Flavones: 50–200 µM22
Galloylated catechins: 0.05–2 µM (exceptionally potent for natural products)23
Why flavonoids matter
They illustrate that reversible, non-covalent scaffolds can be potent if structural rigidity and hydrophobic patch distribution are optimized.
Aurones — Undervalued but Mechanistically Rational Scaffolds
Aurones, with their benzofuranone core, align well with the lipase catalytic groove:
Their carbonyl oxygen orients toward the oxyanion hole.24
The benzofuranone ring stacks against Phe77.4,24
Long alkoxy chains (C6–C10) mimic natural fatty acids and do not trigger covalent chemistry.4,25
Potency
Often 1–10 µM, with synthetic variants dipping <1 µM.25
Why important
Aurones bridge natural and synthetic space: they are rigid, planar, and lipophilic enough to bind well without needing an electrophilic warhead.
Chalcones — Tunable, Conjugated, and Lipase-Compatible
The α,β-unsaturated ketone moiety of chalcones provides:26
Anchor points for hydrogen bonding
A conjugated π system for aromatic stacking
Highly modifiable 2′,4′-OH pattern for binding orientation.26
Space for long-chain hydrophobic tail addition.27
Potency
Ranges from low micromolar to ~0.3 µM for optimized analogs.27,28
Medicinal chemistry insight
Chalcones are ideal for rational scaffold extension into the acyl-binding channel.28
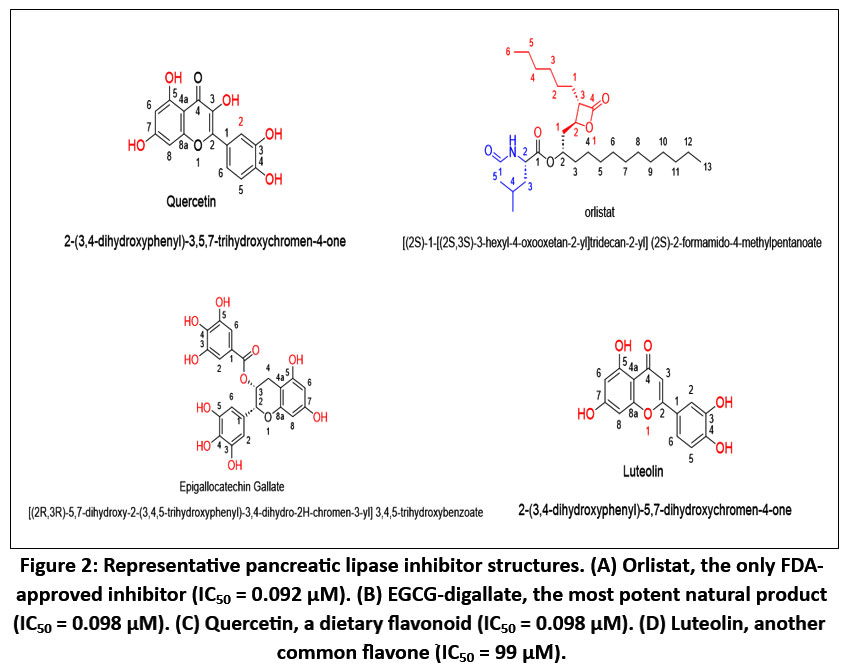 |
Figure 2: Representative pancreatic lipase inhibitor structures. (A) Orlistat, the only FDA-approved inhibitor (IC50 = 0.092 μM). (B) EGCG-digallate, the most potent natural product (IC50 = 0.098 μM).
|
Synthetic Scaffolds — Where Medicinal Chemistry Truly Begins
Thiazolidinedione (TZD) (Reversible Covalent Mimics without the Risks)
TZDs mimic the orientation of the catalytic serine’s transition state but do so reversibly.29 They engage the catalytic pocket by:
Acting as hydrogen-bond acceptors.4
Presenting large, polarizable aromatic “faces” to Phe77.4,30
Aligning substituents to occupy the hydrophobic channel.30
Potency
Often 2–10 µM, with optimized derivatives approaching submicromolar.29
Quinazolinone–Coumarin Hybrids — Dual-Action Precision
These scaffolds introduce:
A quinazolinone core for hydrogen bonding near Ser152
A coumarin chromophore for aromatic stacking.31
Halogenated benzyl groups providing hydrophobic anchoring.32
Unique advantage: simultaneous lipase + α-glucosidase inhibition — ideal for metabolic syndrome.32
Triazoles — Small, Cheap, Stable, and Surprisingly Bioactive
Triazoles
Offer predictable synthetic access (CuAAC “click” chemistry).33
Provide heteroatom-rich surfaces for hydrogen bonding.34
Support bulky aromatic groups that fit the hydrophobic tunnel.35
Potency
Low micromolar (1–5 µM) for optimized variants.36
Unified Structure-Activity Principles Across Inhibitor Classes
Comparative analysis of all inhibitor classes reveals convergent SAR principles that define pancreatic lipase binding affinity:
Hydrophobic Tunnel Engagement
The acyl-binding channel accommodates linear chains of C10-C15 optimal length.4,17 β-Lactones exploit this with saturated fatty acid mimics, while aurones and chalcones achieve equivalent occupancy through extended alkoxy substituents (C6-C10).24,25,27 Chains shorter than C6 lose tunnel contacts; chains beyond C16 induce steric clashes with Ile209 and Val260.4
Aromatic Platform Interactions
Phe77 and Tyr114 form a π-stacking platform essential for inhibitor orientation.21 Flavonoids position their B-ring against this surface, TZDs present thiazolidinedione aromatic faces, and triazoles dock heterocyclic cores.Compounds lacking planar aromatic systems show 10-50 fold potency loss regardless of other favorable features.37,38
Catalytic Triad Recognition
Hydrogen bonding to Ser152 (directly or via water-mediated contacts) and His263 stabilizes the binding pose.38 The oxyanion hole (Phe77, Leu153 backbone NHs) provides additional anchoring for carbonyl or hydroxyl acceptors.EGCG derivatives achieve sub-micromolar potency through simultaneous engagement of all three interaction nodes.24,39
Conformational Compatibility
Rigid, planar scaffolds (aurones, chalcones, quinazolinones) maintain pre-organized conformations that minimize entropic penalties upon binding. Flexible inhibitors pay conformational costs that reduce effective binding free energy by 1-2 kcal/mol.
The integration of these four principles—rather than optimization of any single feature—distinguishes sub-micromolar inhibitors from weak binders across all chemical classes.
Table 1: provides a comprehensive comparison of representative inhibitors across all chemical classes, highlighting IC₅₀ values, mechanisms of action, and key structural determinants of potency.”
Table 1: Representative Pancreatic Lipase Inhibitors
| Class | Compound | IC₅₀ | Mechanism | Key Structural Features |
| β-Lactones | ||||
| Lipstatin 17 | 0.14 μM | Irreversible covalent | β-lactone, C13 unsaturated chain | |
| Orlistat 18 | 0.092 μM | Irreversible covalent | Saturated lipstatin derivative | |
| Galloylated Catechins | ||||
| EGCG-3,5-digallate23 | 0.098 μM | Reversible competitive | Di-galloylated flavan-3-ol | |
| Oolonghomobisflavan A23 | 0.048 μM | Reversible competitive | Dimeric catechin | |
| EGCG23 | 0.349 μM | Reversible competitive | Mono-galloylated | |
| Flavones | ||||
| 4′-Amino baicalein | <1.0 μM | Reversible competitive | Modified pyrogallol core | |
| Myricetin | ~1.5 μM | Reversible non-competitive | Hexahydroxy flavone | |
| Quercetin | 1.5-128 μM | Reversible non-competitive | Pentahydroxy flavone | |
| Luteolin | 99 μM | Reversible competitive | Tetrahydroxy flavone | |
| Biflavones | ||||
| Isoginkgetin 40 | 2.9 μM | Reversible mixed | Ginkgo biflavone | |
| Aurones | ||||
| 4,6-Dialkoxyaurone 23 | 1.95 μM | Reversible | Long alkoxy chains (C6-C10) | |
| Chalcones | ||||
| Compound B1327 | 0.33 μM | Reversible mixed | Two long carbon chains | |
| Sanggenon D 41 | 0.77 μM | Reversible mixed | Diels-Alder adduct | |
| TZD Derivatives | ||||
| Arylidene-TZD (18a)29 | 2.71 μM | Reversible competitive | 2,5-Disubstituted arylidene | |
| Indole-TZD (7k)30 | 7.30 μM | Reversible competitive | Indole-3-carboxaldehyde hybrid | |
| Quinazolinone-Coumarin | ||||
| 4-Bromo derivative 32 | 2.85 μM | Reversible | Dual PL/α-glucosidase | |
| Triazoles | ||||
| p-Fluoro-benzyl 34,36 | 1.1 μM | Reversible | Click chemistry product |
Computational Approaches
Computational tools are uniquely powerful for lipase because multiple high-resolution structures exist in both “open” and “closed” conformations. The enzyme’s deep, solvophobic pocket, its lid mobility, and the colipase interaction surface create a complex landscape ideally suited for in silico exploration.
Docking — What It Actually Reveals
Effective docking reveals:37
Whether the compound can position a hydrophobic chain parallel to the acyl-binding tunnel.4
If its polar motifs can engage Ser152, His263, or oxyanion-hole NHs1,42
How well its aromatics align with Phe77 / Tyr11424,42
Whether bulky substituents clash with the lid domain.4
Docking consistently highlights a “triad interaction cluster” — the positioning of the ligand relative to Ser152, His263, and Phe77 predicts 60–80% of potency variance.
Molecular Dynamics (MD)— Capturing the Enzyme’s Real Behavior
MD reveals:43
Lid flexibility over 50–200 ns44
Stability of hydrogen bonds (Ser152/His263 occupancy %)4,44
Water molecules mediating key contacts45
Shape complementarity changes
Whether bulky ligands distort the tunnel (undesirable)
For potent ligand–enzyme complexes:
Root mean square deviation (RMSD): <3 Å39
Root mean square fluctuation (RMSF) (catalytic residues): <1.5 Å39
H-bond occupancy: >50%45
Hydrophobic tunnel remains undisturbed4
Quantitative Structure-Activity Relationship (QSAR) — What Actually Improves Potency
Across published QSAR models, three descriptor clusters consistently correlate with PL inhibition:
Hydrophobicity
Lipophilic interactions explain why C10–C15 chains outperform others.46
Planarity & rigidity47
Favorable for π–π stacking and entering the narrow tunnel.
H-bond acceptor count
Essential for catalytic triad engagement.38,46
Revised rule-of-thumb:38,47
LogP ~3–6 + planar core + one HBA near Ser15242 → good starting point.
Pharmacophore Models — The “Minimum Binding Requirements”
Consensus pharmacophore typically includes:
1 Hydrogen bond acceptor (HBA) near Ser152.1,42
Two hydrophobic sites along the tunnel1,4
One aromatic ring for stacking42
Optional HBD for oxyanion hole stabilization
Any scaffold lacking the hydrophobic pair almost always shows weak activity.4,48
Artificial Intelligence (AI) & Fragment-Based Drug Design (FBDD) — Modern Tools for a Traditionally Overlooked Target
Machine learning (ML) models rapidly predict lipase inhibitors with >80% classification accuracy.47,49
Generative deep-learning models can propose non–β-lactone scaffolds optimized for potency + GI confinement.50,51
FBDD identifies small tunnel-binding fragments that can be linked or merged to produce potent, lipophilic inhibitors without covalent warheads.52
AI offers a path to lipase inhibitors without the problems of orlistat, a previously unthinkable direction.
Table 2 summarizes validation metrics from key computational studies, demonstrating strong correlations between in silico predictions and experimental potency data, validating these methodologies for prospective inhibitor design.
Table 2: Computational Validation Metrics
| Study | Method | Key Result | Correlation |
| Indole-TZD series 30 | AutoDock + in vitro | 4 H-bonds with Ser152/His263 | r = 0.91 (p<0.05) |
| Arylidene-TZD 18a 29 | Docking + 200ns MD | RMSD 2.8±0.4 Å, 3.2 H-bonds | ΔG = -32.5 kcal/mol |
| Pyrazolyl-TZD 11e 53 | 100ns MD simulation | Stable RMSD ~3 Å, Rg ~20.5 Å | Maintained integrity |
| Aurone derivatives 24 | Docking (82 compounds) | 62/82 engaged catalytic triad | Tunnel occupancy key |
| Chalcone series 27 | Docking validation | Carbonyl H-bonds to Ser152/His263 | R² = 0.986 |
| Flavonoid QSAR 47 | Multi-descriptor model | LogP, OH count, planarity | R² = 0.82, Q² = 0.75 |
Comparative analysis of irreversible versus reversible inhibition strategies reveals distinct trade-offs between potency, tolerability, and therapeutic flexibility (Table 3). While β-lactones achieve superior enzymatic inhibition, reversible scaffolds offer advantages in safety profiles and mechanistic versatility that may prove decisive for next-generation therapeutics.
Table 3: β-Lactone vs. Reversible Inhibitors – Comparative Profile
| Parameter | β-Lactones | Reversible Inhibitors |
| Potency (IC₅₀)18,23,29 | 0.092 μM | 0.048-2.85 μM |
| Mechanism 17,29 | Irreversible covalent | Reversible competitive/mixed |
| GI Tolerability 2,54 | Poor (steatorrhea 15-30%) | Improved (partial inhibition) |
| Systemic Exposure 55,56 | Minimal (<1%) | Variable (0.5-15%) |
| Chemical Stability 3,17,23,29 | High | Moderate |
| Selectivity 3,54 | Moderate | Higher (structure-dependent) |
| Allosteric Potential 54 | None | Possible |
| Multi-Target Capability 32,57 | Limited | High (hybrid scaffolds) |
| Clinical Stage 2,3 | Approved (1999) | Preclinical to Phase I |
Future Medicinal Chemistry Strategies
Allosteric and Partial Inhibition (“Smart Inhibition”)
Instead of knocking out lipase activity entirely, future inhibitors aim for controlled attenuation:
40–70% inhibition.54
Minimal GI side effects
Preserved gastrointestinal physiology
Improved adherence
Reduced compensatory overeating
Targeting the lid hinge, colipase interface, or surface loops provides viable allosteric entry points.4,54,58,59
Multi-Target Chemotypes — Metabolic Network Thinking
Obesity is not a single-pathway disease. Hybrid molecules that act on:32,57
PL + α-glucosidase.32
PL + peroxisome proliferator-activated receptor gamma (PPARγ).57
PL + AMP-activated protein kinase (AMPK)60
can modulate both fat and carbohydrate absorption while influencing systemic metabolism.
Dual-modulators = lower doses, fewer side effects, greater metabolic impact.61
Beyond β-Lactones — Safe Covalent and Non-Covalent Alternatives
Medicinal chemists are now exploring:
Boronic acids (reversible covalent, tunable)62,63
Phenyl carbamates (stable, non-strained electrophiles)64,65
Soft electrophiles activated only near Ser152.66
These promise potency without the toxicity of classic β-lactones.62,64
Bridging the Translational Gap: Why Potent In Vitro Inhibitors Fail Clinically
Despite numerous inhibitors demonstrating sub-micromolar potency in enzymatic assays, only orlistat has achieved clinical approval, revealing critical translational barriers.3 Potent natural products like galloylated catechins (IC₅₀ 0.048 μM) undergo extensive degradation in gastric acid, rapid enterocyte metabolism, and oxidative instability under intestinal conditions, dramatically reducing effective concentrations at the lipid-water interface.23,56Additionally, complete lipase inhibition triggers compensatory physiological responses—pancreatic enzyme hypersecretion, altered bile acid synthesis, and microbiome-mediated fat metabolism—explaining why 95% enzymatic inhibition translates to only 30% reduction in fat absorption clinically.2 Overcoming these barriers requires integrated preclinical models incorporating simulated intestinal conditions and mechanistic biomarkers that predict clinical fat malabsorption beyond simple IC₅₀ values.
Targeting the Lipophilic Tunnel — The Real Key to Potency
New scaffolds explicitly aim to:
Extend into the hydrophobic tunnel.1
Position aromatic or aliphatic substituents to fill “hot spots”.4
Exploit tunnel residues (Ile209, Leu213, Val260, Phe215).67
This is the feature β-lactones exploit — medicinal chemists can mimic it without irreversible chemistry.3
Designing GI-Localized Drugs
Best-in-class PL inhibitors will be:
Highly lipophilic (LogP > 5)55
High molecular weight55
Poorly permeable55,56
Designed for local GI action56
Cleared almost exclusively in feces56,68,69
This pharmacokinetic design is a feature, not a flaw — the exact opposite of classical drug design.
Discussions
This comprehensive review synthesizes mechanistic, structural, and medicinal chemistry perspectives on pancreatic lipase inhibition, revealing both the molecular basis for current therapeutic limitations and the strategic pathways toward improved inhibitor design.
From Mechanism to Medicinal Chemistry: Integrating Biological Insights
The interfacial activation mechanism of pancreatic lipase—unique among digestive enzymes—presents both opportunities and challenges for inhibitor development.1,7 Unlike soluble enzymes where active-site geometry alone dictates binding, lipase requires inhibitors to function at lipid-water interfaces, engage dynamically with the lid domain, and compete with physiological substrates organized in mixed micelles. This explains why classical structure-activity relationship predictions often fail: compounds optimized for aqueous binding may not maintain potency under interfacial conditions.8
The catalytic triad (Ser152-His263-Asp176) and oxyanion hole provide well-defined molecular targets,8,9 yet β-lactone inhibitors—despite near-perfect transition-state mimicry17,18 suffer from irreversible chemistry that triggers compensatory physiological responses.2 This potency-tolerability paradox underscores a critical lesson: maximal enzyme inhibition does not translate to optimal therapeutic outcomes. The future lies in partial, tunable inhibition (40-70% activity reduction)54 that preserves basal lipid digestion while reducing caloric absorption.
Reversible Inhibitors as Safer Alternatives: Balancing Potency and Physiological Compatibility
Natural products, particularly galloylated catechins (IC₅₀ 0.048 μM) and optimized chalcones (IC₅₀ 0.33 μM),27 demonstrate that reversible, non-covalent scaffolds can achieve potencies rivaling orlistat when designed for optimal hydrophobic tunnel occupancy and aromatic platform engagement.24,25 However, their clinical translation is hindered by metabolic instability, pH-dependent degradation, and poor formulation characteristics.23,56
Synthetic scaffolds—thiazolidinediones,29,30 triazoles,34–36 quinazolinone-coumarin hybrids32 address these limitations through enhanced chemical stability and tunable pharmacokinetic properties. The quinazolinone-coumarin dual inhibitors represent a particularly promising strategy: simultaneous modulation of pancreatic lipase and α-glucosidase addresses both fat and carbohydrate absorption, potentially offering superior metabolic control with lower individual enzyme inhibition requirements.
Computational Tools: From Predictive Limitations to Strategic Utility
Molecular docking and dynamics simulations have proven invaluable for understanding binding modes and identifying key interaction hotspots (Ser152, His263, Phe77, Tyr114).37,42,70 However, their predictive accuracy for interfacial enzymes remains constrained by the inability to model lipid-water boundaries, bile salt effects, and colipase-mediated conformational changes. Computational tools should thus be viewed as hypothesis-generating and mechanistic-interrogation instruments rather than definitive predictors of in vivo efficacy.
QSAR models consistently identify lipophilicity (LogP 3-6), planarity, and hydrogen-bond acceptor capacity as dominant determinants of potency,46,47 but this risks reinforcing lipophilic bias that contributes to poor bioavailability and formulation challenges. Machine-learning and AI-driven scaffold generation50–52 offer opportunities to escape this local chemical space, potentially identifying novel non-β-lactone frameworks optimized for both potency and drug-like properties.
Translational Barriers: Why In Vitro Potency Fails Clinically
The persistent disconnect between enzymatic potency and clinical efficacy reflects fundamental misalignments between experimental models and physiological reality.3,56 Simplified assays using p-nitrophenyl substrates bypass interfacial activation requirements, while in vivo fat digestion involves bile salt competition, colipase stabilization, and complex micellar organization.1,8 Furthermore, complete lipase inhibition triggers adaptive responses—pancreatic enzyme hypersecretion, altered bile acid synthesis, microbiome-mediated compensatory pathways—that diminish therapeutic impact despite sustained enzymatic blockade.2
Successful next-generation inhibitors must be designed with these physiological constraints in mind: GI-localized pharmacokinetics (high MW, extreme lipophilicity, poor permeability55,56) to minimize systemic exposure; partial inhibition profiles to avoid triggering compensatory mechanisms; and multi-target activity to address metabolic dysfunction comprehensively rather than through single-enzyme modulation.
Strategic Priorities for Future Inhibitor Development
Four strategic directions emerge from this integrated analysis:
Allosteric and partial inhibition: Targeting the lid-colipase interface or surface loops54,58,71 to achieve controllable, tunable activity reduction without complete active-site blockade.
Multi-target hybrid molecules: Dual PL/α-glucosidase32 or PL/PPARγ57 inhibitors that address multiple metabolic pathways simultaneously, potentially reducing required doses and side-effect burden.
Non-β-lactone electrophiles: Boronic acids, carbamates, and sulfonyl fluorides62–66 offer reversible or slowly-reversible covalent mechanisms with reduced toxicity profiles compared to classical β-lactones.
GI-restricted pharmacokinetics: Deliberate design for intestinal localization55,56 through high molecular weight, extreme lipophilicity, and poor permeability—converting traditional “drug-likeness” liabilities into therapeutic assets.
The ultimate goal is not to replicate orlistat’s potency but to transcend its limitations: achieving meaningful fat malabsorption with improved tolerability, adherence, and long-term sustainability. This requires embracing partial inhibition, allosteric modulation, and multi-target strategies that align molecular design with physiological reality.
Conclusion
Pancreatic lipase represents a structurally well-characterized hydrolytic enzyme whose inhibition offers rational obesity management through reduced dietary fat absorption. However, clinical success has been limited by orlistat’s potency-tolerability paradox: irreversible covalent inhibition triggers gastrointestinal side effects and compensatory physiological responses that diminish long-term efficacy.
This review demonstrates that future progress lies not in replicating orlistat’s mechanism but in transcending it through innovative medicinal chemistry. Reversible natural products—galloylated catechins, aurones, and chalcones—achieve sub-micromolar potencies when optimized for hydrophobic tunnel engagement and catalytic triad recognition. Synthetic scaffolds including thiazolidinediones, triazoles, and quinazolinone-coumarin hybrids extend this principle through enhanced stability, tunable pharmacokinetics, and multi-target capabilities.
Computational approaches combining molecular dynamics, QSAR modeling, and AI-driven scaffold generation provide mechanistic insights that accelerate rational inhibitor development, though interfacial enzyme behavior remains incompletely modeled.
Four strategic priorities define next-generation development: allosteric and partial inhibition targeting the lid-colipase axis; multi-target hybrids addressing both lipid and carbohydrate metabolism; non-β-lactone electrophiles offering safer covalent mechanisms; and GI-restricted pharmacokinetics through deliberate high molecular weight design.
Success requires embracing partial rather than maximal inhibition, allosteric rather than purely active-site targeting, and multi-target rather than single-enzyme modulation. By aligning molecular design with pancreatic lipase’s unique interfacial activation mechanism, next-generation inhibitors can achieve meaningful clinical impact while avoiding orlistat’s tolerability limitations
Acknowledgement
The authors gratefully acknowledge the Almighty for guidance and strength, their families for unwavering support and, Gujarat Technological University, Ahmedabad, for providing the necessary institutional support and facilities that enabled the successful completion of this work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable
Author Contributions
Neil Bienkumar Panchal: Conceptualization, Literature review, Data curation, Writing – Original draft, Visualization, Validation.
Vipul Manusinh Vaghela: Supervision, Writing – Review & editing, Project administration, Final approval. All authors have read and approved the final manuscript for publication.
References
- Van Tilbeurgh H, Egloff MP, Martinez C, Rugani N, Verger R, Cambillau C. Interfacial activation of the lipase–procolipase complex by mixed micelles revealed by X-ray crystallography. Nat 1993 3626423. 1993;362(6423):814-820. doi:10.1038/362814a0
CrossRef - Filippatos TD, Derdemezis CS, Gazi IF, Nakou ES, Mikhailidis DP, Elisaf MS. Orlistat-associated adverse effects and drug interactions: a critical review. Drug Saf. 2008;31(1):53-65. doi:10.2165/00002018-200831010-00005
CrossRef - Lunagariya NA, Patel NK, Jagtap SC, Bhutani KK. Inhibitors of pancreatic lipase: state of the art and clinical perspectives. EXCLI J. 2014;13:897. Accessed November 18, 2025. https://pmc.ncbi.nlm.nih.gov/articles/PMC4464291/
- Egloff MP, Marguet F, Buono G, Verger R, Cambillau C, van Tilbeurgh H. The 2.46 A resolution structure of the pancreatic lipase-colipase complex inhibited by a C11 alkyl phosphonate. Biochemistry. 1995;34(9):2751-2762. doi:10.1021/BI00009A003
CrossRef - Cerchia C, Lavecchia A. New avenues in artificial-intelligence-assisted drug discovery. Drug Discov Today. 2023;28(4):103516. doi:10.1016/J.DRUDIS.2023.103516
CrossRef - Tong X, Liu X, Tan X, et al. Generative Models for De Novo Drug Design. J Med Chem. 2021;64(19):14011-14027. doi:10.1021/ACS.JMEDCHEM.1C00927
CrossRef - Verger R. ‘Interfacial activation’ of lipases: facts and artifacts. Trends Biotechnol. 1997;15(1):32-38. doi:10.1016/S0167-7799(96)10064-0
CrossRef - Van Tilbeurgh H, Sarda L, Verger R, Cambillau C. Structure of the pancreatic lipase–procolipase complex. Nat 1992 3596391. 1992;359(6391):159-162. doi:10.1038/359159a0
CrossRef - Chapus C, Sémériva M. Mechanism of pancreatic lipase action. 2. Catalytic properties of modified lipases. Biochemistry. 1976;15(23):4988-4991. doi:10.1021/BI00668A007
CrossRef - Honeder SE, Tomin T, Schinagl M, et al. Research Advances Through Activity-Based Lipid Hydrolase Profiling. Isr J Chem. 2023;63(3-4):e202200078. doi:10.1002/IJCH.202200078;CSUBTYPE:STRING:SPECIAL;PAGE:STRING:ARTICLE/CHAPTER
CrossRef - Reis P, Miller R, Leser M, Watzke H. Lipase-catalyzed Reactions at Interfaces of Two-phase Systems and Microemulsions. Appl Biochem Biotechnol. 2008;158(3):706. doi:10.1007/S12010-008-8354-5
CrossRef - Van Tilbeurgh H, Bezzine S, Cambillau C, Verger R, Carrière F. Colipase: Structure and interaction with pancreatic lipase. Biochim Biophys Acta – Mol Cell Biol Lipids. 1999;1441(2-3):173-184. doi:10.1016/S1388-1981(99)00149-3
CrossRef - Yang Y, Lowe ME. The open lid mediates pancreatic lipase function. J Lipid Res. 2000;41(1):48-57. doi:10.1016/s0022-2275(20)32073-3
CrossRef - Winkler FK, D’Arcy A, Hunziker W. Structure of human pancreatic lipase. Nat 1990 3436260. 1990;343(6260):771-774. doi:10.1038/343771a0
CrossRef - Lim SY, Steiner JM, Cridge H. Lipases: it’s not just pancreatic lipase! Am J Vet Res. 2022;83(8). doi:10.2460/AJVR.22.03.0048
CrossRef - Hermoso J, Pignol D, Kerfelec B, Crenon I, Chapus C, Fontecilla-Camps JC. Lipase Activation by Nonionic Detergents: THE CRYSTAL STRUCTURE OF THE PORCINE LIPASE-COLIPASE-TETRAETHYLENE GLYCOL MONOOCTYL ETHER COMPLEX. J Biol Chem. 1996;271(30):18007-18016. doi:10.1074/JBC.271.30.18007
CrossRef - Hochuli E, Kupfer E, Maurer R, Meister W, Mercadal Y, Schmidt K. Lipstatin, an inhibitor of pancreatic lipase, produced by Streptomyces toxytricini. II. Chemistry and structure elucidation. J Antibiot (Tokyo). 1987;40(8):1086-1091. doi:10.7164/ANTIBIOTICS.40.1086
CrossRef - Hadvary P, Lengsfeld H, Wolfer H. Inhibition of pancreatic lipase in vitro by the covalent inhibitor tetrahydrolipstatin. Biochem J. 1988;256(2):357-361. doi:10.1042/BJ2560357
CrossRef - Camara K, Kamat SS, Lasota CC, Cravatt BF, Howell AR. Combining cross-metathesis and activity-based protein profiling: New β-lactone motifs for targeting serine hydrolases. Bioorg Med Chem Lett. 2015;25(2):317-321. doi:10.1016/J.BMCL.2014.11.038
CrossRef - Wu X, He W, Yao L, et al. Characterization of Binding Interactions of (−)-Epigallocatechin-3-gallate from Green Tea and Lipase. J Agric Food Chem. 2013;61(37):8829-8835. doi:10.1021/JF401779Z
CrossRef - Park JY, Kim CS, Park KM, Chang PS. Inhibitory characteristics of flavonol-3-O-glycosides from Polygonum aviculare L. (common knotgrass) against porcine pancreatic lipase. Sci Reports 2019 91. 2019;9(1):18080-. doi:10.1038/s41598-019-54546-8
CrossRef - Lee EM, Lee SS, Chung BY, et al. Pancreatic Lipase Inhibition by C-Glycosidic Flavones Isolated from Eremochloa ophiuroides. Mol 2010, Vol 15, Pages 8251-8259. 2010;15(11):8251-8259. doi:10.3390/MOLECULES15118251
CrossRef - Nakai M, Fukui Y, Asami S, et al. Inhibitory effects of oolong tea polyphenols on pancreatic lipase in vitro. J Agric Food Chem. 2005;53(11):4593-4598. doi:10.1021/jf047814+
CrossRef - Nguyen PTV, Huynh HA, van Truong D, Tran TD, Vo CVT. Exploring Aurone Derivatives as Potential Human Pancreatic Lipase Inhibitors through Molecular Docking and Molecular Dynamics Simulations. Molecules. 2020;25(20). doi:10.3390/MOLECULES25204657
CrossRef - Thi Vo C Van, Thanh Nguyen T, Ngoc Dang T, et al. Design, synthesis, biological evaluation and molecular docking of alkoxyaurones as potent pancreatic lipase inhibitors. Bioorg Med Chem Lett. 2024;98:129574. doi:10.1016/J.BMCL.2023.129574
CrossRef - Tam DNH, Mostafa EM, Tu VL, et al. Efficacy of chalcone and xanthine derivatives on lipase inhibition: A systematic review. Chem Biol Drug Des. 2020;95(2):205-214. doi:10.1111/CBDD.13626
CrossRef - Huo PC, Hu Q, Shu S, et al. Design, synthesis and biological evaluation of novel chalcone-like compounds as potent and reversible pancreatic lipase inhibitors. Bioorg Med Chem. 2021;29:115853. doi:10.1016/J.BMC.2020.115853
CrossRef - Jeong JY, Jo YH, Kim SB, et al. Pancreatic lipase inhibitory constituents from Morus alba leaves and optimization for extraction conditions. Bioorganic Med Chem Lett. 2015;25(11):2269-2274. doi:10.1016/j.bmcl.2015.04.045
CrossRef - Dhiman P, Yadav N, Auti PS, et al. Discovery of thiazolidinedione-based pancreatic lipase inhibitors as anti-obesity agents: synthesis, in silico studies and pharmacological investigations. J Biomol Struct Dyn. 2025;43(12):5756-5778. doi:10.1080/07391102.2024.2310799
CrossRef - George G, Auti PS, Paul AT. Design, synthesis, in silico molecular modelling studies and biological evaluation of novel indole-thiazolidinedione hybrid analogues as potential pancreatic lipase inhibitors. New J Chem. 2021;45(3):1381-1394. doi:10.1039/D0NJ05649A
CrossRef - Ibrar A, Shehzadi SA, Saeed F, Khan I. Developing hybrid molecule therapeutics for diverse enzyme inhibitory action: Active role of coumarin-based structural leads in drug discovery. Bioorg Med Chem. 2018;26(13):3731-3762. doi:10.1016/J.BMC.2018.05.042
CrossRef - Menteşe E, Karaali N, Akyüz G, Yılmaz F, Ülker S, Kahveci B. Synthesis and Evaluation of α-Glucosidase and Pancreatic Lipase Inhibition by Quinazolinone-Coumarin Hybrids. Chem Heterocycl Compd 2017 5212. 2017;52(12):1017-1024. doi:10.1007/S10593-017-2002-3
CrossRef - Thirumurugan P, Matosiuk D, Jozwiak K. Click chemistry for drug development and diverse chemical-biology applications. Chem Rev. 2013;113(7):4905-4979. doi:10.1021/CR200409F
CrossRef - Ozdemir Y, Bekircan O, Baltaş N, Menteşe E. Synthesis and pancreatic lipase inhibitory activities of some 1,2,4‐triazol‐5(3)‐one derivatives. J Heterocycl Chem. 2020;57(12):4239-4253. doi:10.1002/JHET.4130
CrossRef - Kahveci B, Yılmaz F, Menteşe E, Ülker S. Design, Synthesis, and Biological Evaluation of Coumarin-Triazole Hybrid Molecules as Potential Antitumor and Pancreatic Lipase Agents. Arch Pharm (Weinheim). 2017;350(8). doi:10.1002/ARDP.201600369
CrossRef - Jalaja R, Leela SG, Valmiki PK, et al. Discovery of Natural Product Derived Labdane Appended Triazoles as Potent Pancreatic Lipase Inhibitors. ACS Med Chem Lett. 2018;9(7):662-666. doi:10.1021/ACSMEDCHEMLETT.8B00109
CrossRef - Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935-949. doi:10.1038/NRD1549
CrossRef - Allal H, Nemdili H, Zerizer MA, Zouchoune B. Molecular structures, chemical descriptors, and pancreatic lipase (1LPB) inhibition by natural products: a DFT investigation and molecular docking prediction. Struct Chem 2023 351. 2023;35(1):223-239. doi:10.1007/S11224-023-02176-2
CrossRef - Nirmale D, S. S. Insight into the interaction of human pancreatic lipase with potential anti-obesity drug, Cetilistat, using a molecular docking and molecular dynamics simulation. TMR Pharmacol Res. 2022;2(3):11. doi:10.53388/pr202202011
CrossRef - Liu PK, Weng ZM, Ge GB, et al. Biflavones from Ginkgo biloba as novel pancreatic lipase inhibitors: Inhibition potentials and mechanism. Int J Biol Macromol. 2018;118(Pt B):2216-2223. doi:10.1016/j.ijbiomac.2018.07.085
CrossRef - Hou XD, Ge GB, Weng ZM, et al. Natural constituents from Cortex Mori Radicis as new pancreatic lipase inhibitors. Bioorg Chem. 2018;80:577-584. doi:10.1016/j.bioorg.2018.07.011
CrossRef - Veeramachaneni GK, Raj KK, Chalasani LM, Bondili JS, Talluri VR. High-throughput virtual screening with e-pharmacophore and molecular simulations study in the designing of pancreatic lipase inhibitors. Drug Des Devel Ther. 2015;9:4397-4412. doi:10.2147/DDDT.S84052
CrossRef - Hollingsworth SA, Dror RO. Molecular Dynamics Simulation for All. Neuron. 2018;99(6):1129-1143. doi:10.1016/j.neuron.2018.08.011
CrossRef - Salo-Ahen OMH, Alanko I, Bhadane R, et al. Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development. Process 2021, Vol 9, Page 71. 2020;9(1):71. doi:10.3390/PR9010071
CrossRef - Rudling A, Orro A, Carlsson J. Prediction of Ordered Water Molecules in Protein Binding Sites from Molecular Dynamics Simulations: The Impact of Ligand Binding on Hydration Networks. J Chem Inf Model. 2018;58(2):350-361. doi:10.1021/ACS.JCIM.7B00520
CrossRef - Li YF, Chang YQ, Deng J, et al. Prediction and evaluation of the lipase inhibitory activities of tea polyphenols with 3D-QSAR models. Sci Reports 2016 61. 2016;6(1):34387-. doi:10.1038/srep34387
CrossRef - Yuan Y, Pan F, Zhu Z, et al. Construction of a QSAR Model Based on Flavonoids and Screening of Natural Pancreatic Lipase Inhibitors. Nutrients. 2023;15(15). doi:10.3390/NU15153489
CrossRef - Zhai Y, Wang K, Yu Z, Zhou S, Fan J. Pancreatic lipase inhibitors: Virtual screening and mechanistic analysis. Int J Biol Macromol. 2025;310:143128. doi:10.1016/J.IJBIOMAC.2025.143128
CrossRef - Liu Y, Pan F, Wang O, et al. QSAR model of pancreatic lipase inhibition by phenolic acids and their derivatives based on machine learning and multi-descriptor strategy. J Agric Food Res. 2023;14:100783. doi:10.1016/J.JAFR.2023.100783
CrossRef - Jiang Y, Zhang G, You J, et al. PocketFlow is a data-and-knowledge-driven structure-based molecular generative model. Nat Mach Intell 2024 63. 2024;6(3):326-337. doi:10.1038/s42256-024-00808-8
CrossRef - Sun H, Li J, Zhang Y, et al. Hot-Spot-Guided Generative Deep Learning for Drug-Like PPI Inhibitor Design. Interdiscip Sci. Published online 2025. doi:10.1007/S12539-025-00756-W
CrossRef - Keseru GM, Erlanson DA, Ferenczy GG, Hann MM, Murray CW, Pickett SD. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J Med Chem. 2016;59(18):8189-8206. doi:10.1021/ACS.JMEDCHEM.6B00197
CrossRef - S.N.C. S, Bhurta D, Kantiwal D, George G, Monga V, Paul AT. Design, synthesis, biological evaluation and molecular modelling studies of novel diaryl substituted pyrazolyl thiazolidinediones as potent pancreatic lipase inhibitors. Bioorg Med Chem Lett. 2017;27(16):3749-3754. doi:10.1016/J.BMCL.2017.06.069
CrossRef - Moreno-Córdova EN, Arvizu-Flores AA, Valenzuela-Soto EM, et al. Gallotannins are uncompetitive inhibitors of pancreatic lipase activity. Biophys Chem. 2020;264. doi:10.1016/j.bpc.2020.106409
CrossRef - Dorel R, Wong AR, Crawford JJ. Trust Your Gut: Strategies and Tactics for Intestinally Restricted Drugs. ACS Med Chem Lett. 2023;14(3):233. doi:10.1021/ACSMEDCHEMLETT.3C00001
CrossRef - Hua S. Advances in Oral Drug Delivery for Regional Targeting in the Gastrointestinal Tract – Influence of Physiological, Pathophysiological and Pharmaceutical Factors. Front Pharmacol. 2020;11:496058. doi:10.3389/FPHAR.2020.00524/FULL
CrossRef - Martin H, McGhie TK, Bentley-Hewitt K, Christeller J. PPARγ as a sensor of lipase activity and a target for the lipase inhibitor orlistat. Lipids Health Dis. 2013;12(1). doi:10.1186/1476-511X-12-48
CrossRef - Zhao Y, Zhang M, Hou X, et al. Design, synthesis and biological evaluation of salicylanilides as novel allosteric inhibitors of human pancreatic lipase. Bioorg Med Chem. 2023;91. doi:10.1016/J.BMC.2023.117413
CrossRef - Khan FI, Lan D, Durrani R, Huan W, Zhao Z, Wang Y. The lid domain in lipases: Structural and functional determinant of enzymatic properties. Front Bioeng Biotechnol. 2017;5(MAR):247303. doi:10.3389/FBIOE.2017.00016/FULL
CrossRef - Saeki K, Hayakawa S, Nakano S, et al. In Vitro and In Silico Studies of the Molecular Interactions of Epigallocatechin-3-O-gallate (EGCG) with Proteins That Explain the Health Benefits of Green Tea. Mol 2018, Vol 23, Page 1295. 2018;23(6):1295. doi:10.3390/MOLECULES23061295
CrossRef - Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251-262. doi:10.1038/NRM3311
CrossRef - Lanier M, Cole DC, Istratiy Y, et al. Repurposing Suzuki Coupling Reagents as a Directed Fragment Library Targeting Serine Hydrolases and Related Enzymes. J Med Chem. 2017;60(12):5209-5215. doi:10.1021/ACS.JMEDCHEM.6B01224/ SUPPL_FILE/JM6B01224_SI_003.CSV
CrossRef - Garner CW. Boronic acid inhibitors of porcine pancreatic lipase. J Biol Chem. 1980;255(11):5064-5068. doi:10.1016/s0021-9258(19)70749-2
CrossRef - Chang JW, Cognetta AB, Niphakis MJ, Cravatt BF. Proteome-wide reactivity profiling identifies diverse carbamate chemotypes tuned for serine hydrolase inhibition. ACS Chem Biol. 2013;8(7):1590-1599. doi:10.1021/CB400261H
CrossRef - Alexander JP, Cravatt BF. Mechanism of Carbamate Inactivation of FAAH: Implications for the Design of Covalent Inhibitors and In Vivo Functional Probes for Enzymes. Chem Biol. 2005;12(11):1179. doi:10.1016/J.CHEMBIOL.2005.08.011
CrossRef - Narayanan A, Jones LH. Sulfonyl fluorides as privileged warheads in chemical biology. Chem Sci. 2015;6(5):2650-2659. doi:10.1039/C5SC00408J
CrossRef - Rocha S, Proença C, Araújo AN, et al. Flavonoids as Potential Modulators of Pancreatic Lipase Catalytic Activity. Pharmaceutics. 2025;17(2):163. doi:10.3390/PHARMACEUTICS17020163/S1
CrossRef - Zhi J, Melia AT, Eggers H, Joly R, Patel IH. Review of limited systemic absorption of orlistat, a lipase inhibitor, in healthy human volunteers. J Clin Pharmacol. 1995;35(11):1103-1108. doi:10.1002/J.1552-4604.1995.TB04034.X
CrossRef - Drug P, Information S, Food IUS. Orlistat ( marketed as Alli and Xenical ) Information. 2016;3500:1-2. Accessed November 18, 2025. https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/orlistat-marketed-alli-and-xenical-information
- Hollingsworth SA, Dror RO. Molecular Dynamics Simulation for All. Neuron. 2018;99(6):1129-1143. doi:10.1016/J.NEURON.2018.08.011
CrossRef - Khan FI, Lan D, Durrani R, Huan W, Zhao Z, Wang Y. The lid domain in lipases: Structural and functional determinant of enzymatic properties. Front Bioeng Biotechnol. 2017;5(MAR):247303. doi:10.3389/FBIOE.2017.00016/FULL
CrossRef
Abbreviations
AI – Artificial Intelligence
AMPK – AMP-Activated Protein Kinase
EGCG – Epigallocatechin Gallate
FBDD – Fragment-Based Drug Design
GI – Gastrointestinal
MD – Molecular Dynamics
MM-GBSA – Molecular Mechanics Generalized Born Surface Area
MM-PBSA – Molecular Mechanics Poisson-Boltzmann Surface Area
PDB – Protein Data Bank
PL – Pancreatic Lipase
PPARγ – Peroxisome Proliferator-Activated Receptor Gamma
QSAR – Quantitative Structure-Activity Relationship
RMSD – Root Mean Square Deviation
RMSF – Root Mean Square Fluctuation
SAR – Structure-Activity Relationship
TZD – Thiazolidinedione
Accepted on: 02-02-2026
Second Review by: Dr. Ramya Sri
Final Approval by: Dr. Eugene A. Silow








![]()
![]()
