
Risk- Based Validation of Software, Automation and Artificial intelligence in Pharmaceuticals
 and Manish Dyaneshwar Baviskar2
and Manish Dyaneshwar Baviskar2 1MET’s Institute of Pharmacy, Bhujbal Knowledge City, Adgaon, Nashik, Maharashtra, India.
2Tri-Pac, Inc., South Bend, Indiana, USA
Corresponding Author E-mail:sandeeps_iop@bkc.met.edu
DOI : http://dx.doi.org/10.13005/bbra/3441
Download this article as:
![]()
Artificial intelligence (AI) and machine learning (ML) are transforming the pharmaceutical value chain, yet their adaptive, data-driven behaviour challenges traditional validation frameworks designed for deterministic software. This narrative review synthesizes current global guidance—including GAMP 5 (Second Edition), ALCOA++, ICH Q8–Q11, the U.S. FDA’s Software as a Medical Device (SaMD) AI/ML Action Plan and the 2025 Draft Guidance on Predetermined Change Control Plans for AI/ML-enabled Devices, the European Union AI Act, and ISO/IEC 25010, 27001, and 42001—into a unified, risk-based blueprint encompassing three technology classes: conventional software, automation platforms, and AI/ML models. Differences in user requirement specification, qualification (IQ/OQ/PQ), change management, and lifecycle oversight are mapped, and four emerging pain points—model drift, bias, explainability, and cybersecurity—are highlighted. Building upon the classical V-model, a four-phase roadmap is proposed that integrates deterministic validation discipline with agile, data-centric controls and continuous performance monitoring. Adoption of this blueprint can shorten validation cycles, enhance regulatory compliance, and expedite the delivery of safer and more reliable medicines.
KEYWORDS:Artificial Intelligence; Machine Learning; Risk-Based Validation; Good Manufacturing Practice; Pharmaceutical Preparations; Software Validation; Quality Control; ISO/IEC 42001
Introduction
The pharmaceutical industry is entering a transformative phase with the widespread adoption of Artificial Intelligence (AI) and Machine Learning (ML) technologies across drug development, manufacturing, quality assurance, and regulatory submission landscape.1 These intelligent systems are now employed for predictive analytics, visual inspection, electronic batch record review, and pharmacovigilance decision support, among other critical functions.2 While such technologies offer substantial promise in enhancing operational efficiency and accuracy, they also introduce novel risks and uncertainties particularly around system validation, data integrity, algorithm explainability, and continuous performance monitoring.7
Historically, validation in regulated pharmaceutical environments has been grounded in deterministic logic and static system behavior. Legacy computerized systems were evaluated using frameworks such as GAMP 5, ISO/IEC 25010, and FDA’s 21 CFR Part 11, where qualification protocols (IQ/OQ/PQ) provided a structured approach for system acceptance. However, AI/ML systems challenge these paradigms due to their probabilistic behavior, dynamic learning capabilities, and adaptive decision-making processes. Such complexity necessitates a fundamental shift in validation approaches from static to continuous, from code-centric to data-centric, and from retrospective to proactive lifecycle oversight.
In response to these needs, regulatory bodies have published several pivotal guidance documents in 2025 to address the growing influence of AI. On January 6, 2025, the U.S. Food and Drug Administration (FDA) released the draft guidance titled “Considerations for the Use of Artificial Intelligence ToSupport Regulatory Decision-Making for Drug and Biological Products” which outlines expectations for lifecycle management, change protocols, training dataset documentation, and model transparency for software deployed in regulated settings. 5 A complementary document issued on January 7, 2025” introduces a credibility framework that includes defining context of use (COU), assessing risk, and monitoring post-deployment performance. 6
In parallel, the International Society for Pharmaceutical Engineering (ISPE) updated its updates encourage the adoption of agile validation models, adaptive monitoring strategies, and automated documentation pipelines for AI systems operating within GxP environments. 7
On the global stage, the ratified in late 2024 and effective as of August 2025 classifies AI systems used in pharmaceutical manufacturing, clinical decision support, and patient risk profiling as “high-risk.” The Act mandates conformity assessments, continuous performance monitoring, explainability safeguards, and human oversight controls for such systems.8 In addition, the EMA’s updated Reflection Paper on AI (2024). 9
This review synthesizes these evolving global guidelines and recent industry practices into a comparative roadmap for validation. It aims to bridge traditional software validation techniques with contemporary AI/ML lifecycle governance using a risk-based approach. By comparing validation strategies for deterministic systems, automation platforms, and adaptive AI tools, we propose a harmonized framework suitable for regulatory-compliant integration of intelligent technologies across the pharmaceutical value chain.
Evolution of Pharmaceutical Validation Practices
Validation in the pharmaceutical sector has undergone substantial transformation, evolving from manual documentation methods to complex, risk-based computerized frameworks tailored to ensure system quality, data integrity, and regulatory compliance. Initially, pharmaceutical operations relied on paper-based batch records and retrospective documentation reviews to maintain Good Manufacturing Practice (GMP) standards. However, with the proliferation of digital technologies in laboratory and manufacturing environments, regulatory authorities introduced comprehensive guidelines to ensure the credibility and traceability of electronic systems.
A major regulatory milestone was the introduction of the U.S. Food and Drug Administration’s (FDA) 21 CFR Part 11 in 1997, which established enforceable requirements for electronic records and electronic signatures in GxP-regulated environments. This regulation mandated access controls, audit trails, record retention, and system validation to ensure trustworthy electronic data handling (FDA, 1997).10 In parallel, the European Medicines Agency (EMA) released Annex 11 of the EU GMP guidelines, providing expectations for the validation, documentation, and lifecycle management of computerized systems across European operations.11
As computerized systems became increasingly central to pharmaceutical manufacturing, industry stakeholders recognized the need for structured validation practices. This led to the development of the GAMP® (Good Automated Manufacturing Practice) framework by the International Society for Pharmaceutical Engineering (ISPE). GAMP® 5, first published in 2008 and revised in 2022, advocates for scalable, risk-based validation aligned with system complexity and potential patient safety impact. It provides guidance for developing qualification protocols (Installation Qualification, Operational Qualification, and Performance Qualification), traceability matrices, and risk assessments appropriate to the intended system use.1
In response to growing digitalization, ISPE released an updated GAMP Good Practice Guide: Digital Validation in 2025, addressing the application of validation principles to artificial intelligence (AI), open-source platforms, and decentralized technologies. The guide emphasizes lifecycle monitoring, automated documentation, and hybrid validation strategies suitable for dynamic and adaptive systems operating in GxP environments.7
As these modern validation approaches are increasingly applied to digital technologies, they inherently reinforce the critical role of data integrity in ensuring the reliability and compliance of electronic systems.
Complementing these validation frameworks, data integrity has become a cornerstone of compliance. Originally framed under the ALCOA principles Attributable, Legible, Contemporaneous, Original, Accurate the concept was later expanded to ALCOA++, adding Completeness, Consistency, Endurance, and Availability to better address the nuances of electronic data and complex system architectures10,13. ALCOA++ has since been universally accepted across regulatory agencies, reinforcing that data integrity must be maintained not only during data capture but throughout the lifecycle of data processing, review, and archival.
Recent draft guidance from the FDA in 2025 have reinforced that ALCOA++ principles also apply to AI/ML-enabled systems. Specifically, the FDA’s draft guidance on AI-enabled device software functions outlines expectations for traceability of training data, auditability of algorithm evolution, and lifecycle monitoring of AI system performance. 5 Similarly, in its guidance on the use of AI in regulatory decision-making for drugs and biologics, the FDA emphasizes the importance of predefined risk assessments, context-of-use specificity, and documentation of model behavior under real-world conditions.6
At the international level, regulatory harmonization efforts are also advancing. The European Committee for Standardization published CWA 18211:2025, a reference architecture for trustworthy AI in regulated industrial applications. This framework integrates human oversight, lifecycle documentation, and explainability requirements for AI systems deployed in high-risk environments, such as pharmaceutical manufacturing and clinical research.14 The EMA’s 2024 Reflection Paper on the Use of AI further underscores the need for model reproducibility, dataset curation, and performance transparency throughout the medicinal product lifecycle.9
Collectively, these developments demonstrate a clear trend toward integrating lifecycle validation, real-time monitoring, and robust data governance across all system types from traditional software to adaptive AI models. While legacy principles such as GAMP 5 and ALCOA++ remain vital, their effective application to AI/ML technologies require enhanced traceability, dynamic risk management, and hybrid validation frameworks that can accommodate algorithm evolution. This synthesis of traditional and modern approaches forms the foundation for intelligent, compliant, and future-proof validation practices in the pharmaceutical industry.
The historical development of validation principles in the pharmaceutical domain reflects the industry’s shift from deterministic systems to adaptive technologies. From the introduction of FDA’s 21 CFR Part 11 to recent guidance specific to AI/ML, each milestone has shaped how validation is defined and enforced. Figure 1 reflects a chronological overview of these key milestones, illustrating how foundational regulations have evolved to address growing digital complexity and support the governance of intelligent systems.
Regulatory and Standards Landscape
The regulatory and standards environment shaping validation practices for software, computerized systems, and AI/ML in pharmaceuticals is increasingly comprehensive, harmonizing formal guidance with technical standards across multiple domains. These frameworks ensure systems are robust, secure, and well-governed across their lifecycle.
A cornerstone is the FDA SaMD AI/ML Action Plan (2021), which outlines five key pillars predetermined change control plans, Good Machine Learning Practices (GMLP), transparency, performance monitoring, and global harmonization. It supports Predetermined Change Control Plans (PCCPs) that enable controlled model updates post-market without requiring complete revalidation cycles.4
 |
Figure 1: Timeline of Key Validation Milestones in Pharmaceutical Regulation |
Expanding on this, the FDA’s 2025 draft guidance documents AI/MLEnabled Device Software Functions and AI in Drug and Biological Product Decision Making, focus on credibility, context-of-use, risk assessment, and lifecycle performance oversight, reinforcing the SaMD roadmap for drug-related AI systems.5, 6
In the European sphere, the EMA’s Reflection Paper8,9 on Adaptive AI Algorithms mandates transparency, traceability, and reproducibility for models used throughout the medicinal product lifecycle, including manufacturing, clinical studies, and post-market oversight. These principles align with the pending EU AI Act, which classifies high-risk AI used in drug and process development and require human oversight, risk evaluation, and conformity assessment.
Complementing regulatory initiatives, the ICH Q8–Q11 suite (2023–2024) continues to serve as the quality backbone for pharmaceutical development and lifecycle management. ICH Q817 emphasizes design spaces and critical quality attributes, ICH Q9 centers on quality risk management with explicit relevance to AI/ML and ICH Q10–Q1118,19 underscore system robustness, continuous improvement, and manufacturability governance.
Across technical standards, ISO/IEC 25010 (2011) defines critical software quality attributes e.g., functionality, reliability, and security that apply to both traditional and AI-enabled systems. ISO/IEC 27001 (2022) addresses information security, vital for compliance with data privacy and cybersecurity obligations such as GDPR and HIPAA. Most notably, ISO/IEC 42001 (2023) introduces the first AI Management System Standard (AIMS), focusing on lifecycle governance, transparent documentation, algorithmic fairness, and risk-based control mechanisms. It has been adopted globally, with accredited certifications issued as recently as mid-2025.12,16
Recent peer-reviewed analysis reinforces the criticality of this integrated framework. Niazi (2025) conducted a comprehensive review of regulatory expectations for AI/ML in GMP environments, highlighting alignment between FDA, EMA, and MHRA guidance and advocating for lifecycle, risk-based validation.15 Another high-impact publication in Pharmaceutical Sciences (May 2025) emphasizes governance frameworks, cybersecurity, and continuous monitoring as essential pillars of AI validation in pharma.2
Together, these regulatory and standards frameworks form a multi-layered validation architecture from traditional quality systems to AI-specific oversight ensuring that software, automated systems, and AI/ML models are validated effectively, securely, and compliantly throughout their operational life. As regulatory agencies adapt to the rapid evolution of intelligent systems, a broad set of guidelines and technical standards has emerged to ensure safety, reliability, and traceability throughout the system lifecycle. These documents vary in scope from early frameworks addressing electronic records to recent standards focused on algorithmic fairness and continuous monitoring. To help contextualize these developments, Table 1 organizes the most influential regulatory and standards documents relevant to AI/ML validation in pharmaceuticals. The table outlines the issuing authority, year of release, and key focus areas, offering a concise reference point for professionals navigating the complex regulatory environment surrounding digital systems in GxP settings.
Table 1: Key Regulatory Guidelines and Standards for AI/ML Validation in Pharmaceuticals
| Guideline / Standard | Issuing Body | Year | Focus Area |
| 21 CFR Part 11 | FDA | 1997 | Electronic records and signatures |
| Annex 11 | EMA | 2011 | Computerized system validation |
| GAMP® 5 (2nd Edition) | ISPE | 2022 | Risk-based validation for software & automation |
| SaMD AI/ML Action Plan | FDA | 2021 | AI lifecycle, performance monitoring, transparency |
| AI in Drug Decision-Making (Draft Guidance) | FDA | 2025 | Credibility, context of use, continuous monitoring |
| AI Reflection Paper | EMA | 2024 | Model interpretability, dataset traceability |
| ISO/IEC 25010 | ISO | 2011 | Software quality metrics |
| ISO/IEC 27001 | ISO | 2022 | Information security and privacy |
| ISO/IEC 42001 | ISO | 2023 | AI management system: fairness, traceability |
| CWA 18211 | CEN-CENELEC | 2025 | Trustworthy AI architecture for regulated environments |
System-Specific Validation Approaches
Validation strategies differ markedly across traditional software, automation systems, and AI/ML-based technologies due to their inherent design logic, data dependencies, and behavior over time. Tailoring validation to the characteristics of each system type is critical for compliance, quality assurance, and audit readiness within regulated pharmaceutical environments. A one-size-fits-all validation model is no longer adequate, particularly as AI and intelligent automation redefine system complexity and adaptability.
Different categories of computerized systems used in the pharmaceutical industry such as traditional software, automation platforms, and AI-based technologies require validation strategies that reflect their technical design and regulatory risk. While conventional software systems operate through fixed, rule-based logic, AI/ML models rely on changing data patterns and adaptive behavior, often complicating standard validation methods. Automation systems, which control equipment through integrated hardware and software, require an additional layer of oversight. These contrasts are reflected in Table 2, which provides a side-by-side comparison of validation expectations across these system types, including how they differ in user requirements, qualification activities, risk evaluation, change control, and monitoring throughout the system’s lifecycle.
Table 2: Comparison of Validation Requirements across Traditional Software, Automation Systems, and AI/ML Technologies.
| Validation Component | Traditional Software Systems | Automation Systems | AI/ML Systems |
| User Requirements (URS) | Static; defined functions and workflows | Includes hardware logic, interlocks, control sequences | Goal-based; includes learning objectives, data quality |
| Installation Qualification (IQ) | System setup and environment check | Hardware and network configuration | Environment setup, data pipeline validation |
| Operational Qualification (OQ) | Functional requirement testing | Alarm checks, batch sequencing, sensor inputs | Algorithm verification, training/testing benchmark |
| Performance Qualification (PQ) | Application-specific workflows | Simulated production scenarios | Model generalization, real-world performance |
| Risk Management | Known failure modes (e.g., input/output, access) | Includes mechanical and control-loop risks | Bias, drift, fairness, transparency |
| Change Control | Software versioning | Config changes across HMI, PLC, SCADA | Model updates, retraining, feature changes |
| Lifecycle Monitoring | Periodic review | Maintenance logs, software upgrades | Continuous monitoring, drift detection tools (MLflow, etc.) |
Traditional software systems operate on static logic and deterministic outputs, where the same input consistently yields the same result. These systems include business applications, configuration software, and quality systems with predefined functionality. Validation in this domain typically follows the classical V-model framework, which aligns user requirement specifications (URS) with verification and qualification protocols Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ). Each requirement is mapped to corresponding test cases in a traceability matrix to ensure full coverage and auditability. Regulatory frameworks such as FDA 21 CFR Part 1110, EU GMP Annex 1111, and ISO/IEC 2501012 provide clear guidance on system functionality, security, maintainability, and audit trail requirements.
In contrast, automation systems, such as Distributed Control Systems (DCS), Supervisory Control and Data Acquisition (SCADA), and Programmable Logic Controllers (PLC), involve real-time control of physical processes. These systems interface directly with manufacturing equipment and environmental controls, combining software logic with hardware sequencing. Validation of automation platforms must address both hardware qualification and software configuration, including interlocks, sensor feedback, and control loop logic. Standards such as ISA-88 for batch process control and ISA-95 for enterprise integration provide the architectural basis for sequencing, recipe control, and event-based execution. Qualification must demonstrate alarm functionality, batch execution accuracy, and real-time response integrity across all integrated components. For regulated facilities, this includes ensuring data integrity across Human-Machine Interface (HMI) systems, PLC code modules, and MES interactions.1,18
The most complex validation challenges emerge in AI/ML-based systems, which do not follow deterministic execution paths but instead rely on probabilistic models and data-driven learning. These systems evolve over time as new data is introduced, and outputs may vary even with similar inputs due to model training variability. This non-deterministic behavior disrupts traditional validation structures that assume static codebases and consistent outputs. Instead, AI validation must focus on model reproducibility, explainability, and performance transparency across datasets. Tools like SHAP and LIME support post-hoc interpretability, while ISO/IEC 42001 mandates the traceability of training data, model configuration, version control, and retraining conditions.12
Regulatory guidance from the FDA (2025) emphasizes that AI/ML systems must include predetermined retraining triggers, continuous monitoring pipelines, and documented risk mitigation strategies, particularly in contexts where clinical or quality-critical decisions are made. AI validation packages must capture not only source code and test cases but also model lineage, bias audits, validation/testing datasets, performance metrics, and retraining governance. Revalidation is required when models are updated, inputsshift, or new data patterns emerge. Lifecycle monitoring using tools like MLflow, Neptune.ai, or Amazon SageMaker Model Monitor supports drift detection and performance regression analysis, as recommended by the EMA and WHO. 9,17
As these system types increasingly converge for example, AI modules embedded within automation platforms hybrid validation strategies must be employed. This includes layered risk assessments, dynamic traceability matrices, and combined IQ/OQ/PQ and model validation workflows. Pharmaceutical manufacturers must integrate governance frameworks that accommodate traditional GAMP-based validation and modern ML compliance practices, ensuring end-to-end system control, traceability, and audit readiness.
Comparative Validation Framework
Validation in pharmaceutical environments must be tailored to the unique characteristics and regulatory risks associated with different types of technology. Traditional software systems, automation platforms, and AI/ML models each exhibit distinct lifecycle behaviors, logic architectures, and risk profiles. Consequently, validation frameworks must adopt differentiated strategies across critical validation stages ranging from requirement definition to performance qualification, risk assessment, and lifecycle oversight. This section presents a structured comparison of these approaches to guide validation professionals in applying appropriate controls.
In traditional software systems, validation typically begins with a static user requirements specification (URS) that outlines defined functionalities and workflows. The classic V-model applies here, with structured IQ/OQ/PQ protocols ensuring alignment between user expectations and system behavior. Risk assessments focus primarily on known failure modes such as input validation errors, access control weaknesses, or database connectivity issues and change control is managed through software versioning and audit trails.1,12
In automation systems, the URS includes both hardware dependencies and logic control flows, especially where Programmable Logic Controllers (PLCs), SCADA platforms, or sensor-driven batch execution are involved. Validation must confirm not only software configuration but also the physical response of interconnected components. IQ covers hardware installation and network configuration; OQ verifies alarm response, interlocks, and signal feedback; PQ evaluates system performance under simulated production scenarios. Risk assessments expand to include mechanical and operational hazards, with change control requiring synchronized updates across hardware modules and control software.8,18
AI/ML systems present a paradigm shift. URS for intelligent systems must define goal-oriented behavior, such as predictive accuracy, classification boundaries, or adaptive control objectives. These requirements are data-sensitive and evolve alongside model training and deployment contexts. IQ and OQ in AI validation must incorporate dataset traceability, algorithm verification, and training/testing performance benchmarks. PQ requires monitoring of the model’s ability to generalize from data and sustain performance across changing conditions.
Risk management in AI/ML is particularly complex. Instead of predefined failure modes, risks stem from model drift, algorithmic bias, data imbalance, or overfitting, all of which can undermine reliability. Regulatory frameworks such as ISO/IEC 42001 and the FDA’s 2025 draft guidance emphasize the use of continuous monitoring tools, fairness auditing, and pre-established retraining triggers as critical elements of validation in AI systems.6,12,14
Change control in AI/ML differs fundamentally from traditional software. Here, changes may not stem from code edits but from updates to training data, feature engineering, or retraining schedules. Each data-induced change can result in modified system behavior, necessitating robust documentation, performance testing, and QA approval before release. Emerging best practices recommend integrating model cards, dataset logs, and version-controlled artifacts into the change management workflow.6,17
Lifecycle monitoring in AI must also transition from periodic updates to continuous validation pipelines. Tools such as MLflow, Evidently AI, and SageMaker Model Monitor enable real-time tracking of inference quality, concept drift, and retraining efficacy. These tools complement the pharmaceutical industry’s broader adoption of digital quality management systems (QMS), bringing AI into the fold of regulated continuous improvement systems.
By clearly distinguishing the validation expectations and strategies for software, automation, and AI/ML technologies, organizations can design, fit-for-purpose validation plans aligned with both system complexity and regulatory scrutiny. This approach ensures not only compliance but also operational resilience and data integrity across a digitally evolving pharmaceutical ecosystem.
Figure 2 complements this comparative analysis by visually synthesizing the core validation elements shared across system types, while also highlighting domain-specific requirements essential for a harmonized, risk-based validation strategy.
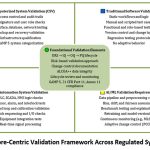 |
Figure 2: Core-Centric Validation Framework Across Regulated System Types |
Key Challenges in AI/ML Validation
While AI and Machine Learning (ML) technologies offer transformative potential in pharmaceutical development, manufacturing, and regulatory decision-making, their validation presents a unique set of challenges that depart significantly from those associated with traditional software or automation systems. These challenges are rooted in the non-deterministic, data-dependent nature of AI, which complicates the application of established validation frameworks. As AI continues to permeate GxP processes, addressing these limitations is essential to ensure compliance, reliability, and patient safety. These validation challenges stem from the inherent unpredictability and learning behavior of AI systems. Figure 3 provides a visual breakdown of these issues, such as model opacity, dataset bias, and algorithmic drift, and maps them to emerging regulatory expectations and mitigation tools like explainability frameworks and monitoring platforms.
 |
Figure 3: Key Challenges in Validating AI/ML in GMP Environments and Corresponding Mitigation Strategies |
One of the most significant obstacles in AI/ML validation is the “black-box” nature of many algorithms, particularly deep learning models. These models often lack inherent explainability, making it difficult for regulators and quality assurance teams to understand how decisions are derived. While tools such as SHAP (SHapley Additive exPlanations), LIME (Local Interpretable Model-Agnostic Explanations), and Integrated Gradients have emerged to provide post hoc interpretability, they often fall short in offering full transparency, particularly when decisions involve life-critical or regulatory endpoints. 6,14 The FDA and EMA now recommend integration of explainability-by-design as part of the AI development lifecycle, especially in high-risk applications such as product release, clinical diagnosis, or pharmacovigilance signal detection.6,9
A second major concern is bias in training datasets, which can lead to systemic fairness issues and performance inconsistencies across populations or product conditions. Pharmaceutical applications often involve imbalanced or non-representative datasets e.g., skewed patient demographics or sensor data inconsistencies which can result in biased predictions or model failures. Regulators now expect sponsors to demonstrate not only statistical accuracy but also fairness, robustness, and equity across populations. Standards such as ISO/IEC TR 24029 and AI ethics frameworks from WHO and OECD advocate for integrated bias detection, impact assessments, and mitigation plans in all AI/ML validation protocols.9,17,20
In contrast to traditional systems that follow predictable logic, AI systems exhibit dynamic behavior over time, adjusting outputs based on new training data, user interaction, or feedback loops. This introduces the risk of model drift, where the performance of the model degrades or shifts from its validated state due to changes in data distribution or environmental context. Tools such as MLflow, SageMaker Model Monitor, and Evidently AI are increasingly being used to track key performance indicators (KPIs) and drift metrics in real-time. However, implementing continuous validation pipelines that maintain regulatory compliance remains a significant challenge particularly when drift occurs slowly or is difficult to detect.6,14
Ensuring compliance with ALCOA++ data integrity principles Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available is also difficult in the context of AI/ML. Model training often involves multiple preprocessing pipelines, external data sources, and non-linear optimization processes. Capturing versioned logs of training data, hyperparameters, intermediate checkpoints, and inference outcomes in a traceable and immutable format is essential but technically demanding. While data versioning tools like DVC and lineage platforms like Neptune.ai offer partial solutions, no unified framework currently exists to enforce ALCOA++ compliance across AI development pipelines in the way that electronic batch records or LIMS systems do for traditional data.10,13
Compounding these technical challenges is a regulatory lag. Despite recent draft guidance, most current GxP validation SOPs and pharmaceutical quality systems are not equipped to handle adaptive, learning-based systems. Many organizations lack AI-specific SOPs, validation templates, or retraining control procedures, which leads to uncertainty during audits and regulatory submissions. International harmonization efforts, such as the FDA’s PCCP model, EMA’s AI oversight principles, and ISO/IEC 42001, are helping bridge these gaps but full integration of AI validation into pharmaceutical quality systems remains a work in progress10,13.
As AI continues to scale across the pharmaceutical value chain, resolving these challenges will be central to its safe and effective adoption. Regulatory bodies, industry stakeholders, and standards organizations must collaborate to co-develop guidance, SOPs, and technology-neutral frameworks that enable both innovation and compliance.
Discussion
The pharmaceutical industry’s transition from deterministic systems to adaptive, data-driven technologies marks a pivotal evolution in system validation. As AI/ML systems become embedded across the pharmaceutical value chain from automated quality control to personalized medicine the foundational assumptions behind validation, traceability, and system oversight are being fundamentally challenged. This discussion synthesizes emerging trends, identifies gaps in current literature and practice, examines regulatory inconsistencies, and proposes future directions for building a robust AI/ML validation ecosystem.
Emerging Trends
A clear trend is the gradual but significant shift from deterministic to adaptive systems within GMP-regulated environments. Traditional rule-based software is increasingly supplemented or even replaced by intelligent systems capable of predictive analytics, anomaly detection, and real-time decision-making. This shift is seen in applications ranging from AI-enhanced visual inspection and real-time release testing to supply chain optimization and adaptive clinical trial designs.15,17
As new use cases emerge, the validation process must adapt to a broader set of system behaviors and performance indicators. Figure 2 highlights several key trends driving this transformation, including the transition from static to adaptive validation, the need for integrated cybersecurity practices, and the growing importance of real-time model monitoring and dynamic SOPs within GMP environments
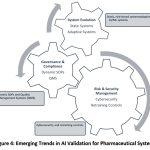 |
Figure 4: Emerging Trends in AI Validation for Pharmaceutical Systems |
As AI adoption increases, so too does the emphasis on data governance and cybersecurity. High-impact standards such as ISO/IEC 27001 and ISO/IEC 42001 reinforce the need for structured data management policies, access controls, and AI lifecycle documentation. Cybersecurity risks associated with externally trained models, third-party datasets, or opaque black-box algorithms have prompted regulatory bodies to issue preliminary guidance on AI security posture and traceability.15,17
However, while regulators are making strides in updating their frameworks such as the FDA’s PCCP model and EMA’s AI reflection papers their pace often lags behind the technical advancements being implemented in industry. Many organizations face uncertainty when validating AI systems that continuously learn or adapt to real-time feedback, especially when legacy SOPs and QMS templates remain static and code-centric.5,6,9
Gaps in Literature and Practice
Despite an increasing body of literature on AI ethics, fairness, and bias mitigation, there remains a notable scarcity of peer-reviewed case studies specifically focused on AI/ML validation within GMP settings. Most industry applications remain either confidential or described in high-level terms, making it difficult to generalize best practices for regulators or practitioners.
Moreover, standardized protocols for model retraining, including revalidation triggers, dataset versioning, and performance benchmarks, are largely undeveloped. Existing quality systems are not designed to manage dynamic, data-driven behaviors, which leads to inconsistent documentation and oversight when AI/ML systems are deployed in live production settings.
The growing use of hybrid systems where AI modules are integrated with traditional automation platforms (e.g., SCADA, MES, PLCs) adds complexity. These hybrid architectures blur the line between software configuration and model inference, challenging conventional IQ/OQ/PQ boundaries and requiring cross-functional validation teams that combine IT, OT, and data science expertise.
Conflicts in Guidance and Interpretation
Another major obstacle to harmonized validation is the divergence in regulatory interpretation between international bodies. For instance, the FDA’s approach to continuous learning systems through PCCPs permits a degree of model evolution within predefined bounds, whereas the EMA emphasizes fixed algorithm configurations with minimal post-deployment change.5,9 These contrasting positions create confusion for global manufacturers attempting to maintain uniform compliance across multiple jurisdictions.
Additionally, while ISO/IEC standards on AI governance (e.g., 42001) and information security (e.g., 27001) are technologically comprehensive, they are not always fully aligned with ICH Q8–Q11 or GAMP guidance, particularly in defining qualification activities or risk acceptance thresholds. This lack of harmonization between standards bodies limits the operationalization of AI validation in quality-driven environments.
There is also a growing debate on whether current PQ (Performance Qualification) frameworks are sufficient for AI systems that learn, adapt, or drift over time. Classical PQ relies on repeatable performance under known inputs, while AI requires dynamic metrics, real-time dashboards, and statistical validation across changing datasets. This philosophical divergence has yet to be resolved in official guidance documents or industry templates.
Future Directions
To address these issues, several forward-looking strategies are gaining traction. First, the development of AI-specific User Requirements Specification (URS) templates that include retraining logic, dataset lineage, and model governance is critical. These templates would define intended use cases, risk scenarios, and lifecycle expectations in ways that align with both ALCOA++ and ISO 42001 standards.
Second, the expansion of GAMP 5 guidance including a potential Appendix specific to AI/ML could provide industry-standard workflows for model onboarding, qualification, and ongoing performance monitoring. To bridge the gap between evolving AI technologies and regulatory requirements, Figure 4 proposes a four-phase roadmap for AI/ML validation in pharmaceutical settings.
 |
Figure 5: Strategic Roadmap for Harmonized AI/ML Validation in Pharma |
It integrates AI-specific requirement templates, hybrid validation workflows, continuous monitoring tools, and global harmonization efforts laying a foundation for scalable and compliant AI deployment in regulated environments. Discussions within ISPE communities suggest increasing support for such an annex, especially for hybrid validation scenarios.
Third, collaborative initiatives such as joint task forces between the FDA, EMA, and international standards bodies could accelerate convergence on AI validation practices. These efforts could mirror past successes with harmonizing quality metrics under ICH and would help reduce fragmentation across geographies.
Finally, the industry must invest in Explainable AI (XAI) technologies and validation-focused ML tools that bridge the gap between data science and regulatory documentation. Emerging platforms such as Evidently AI, Neptune.ai, and Microsoft Responsible AI dashboard provide explainability, drift detection, and version tracking features that align with compliance goals and regulatory expectations.
By recognizing these challenges and proactively addressing the gaps, the pharmaceutical industry can develop a validation paradigm that is both innovation-friendly and regulation-ready, ensuring that AI/ML systems are implemented with safety, reliability, and traceability at their core.
Conclusion and Strategic Recommendations
As pharmaceutical systems grow increasingly complex incorporating adaptive algorithms, real-time automation, and hybrid digital platforms traditional validation methodologies must evolve to remain relevant and effective. The classical frameworks that once guided the qualification of deterministic software and hardware systems are now being stress-tested by the rapid adoption of Artificial Intelligence (AI) and Machine Learning (ML) technologies. These developments demand not only technical innovation but also strategic adaptation across validation, quality assurance, and regulatory compliance domains.
Validation strategies must align with the inherent complexity and behavior of the system in question. For AI/ML-based systems, this means moving beyond static test scripts toward dynamic validation frameworks that incorporate lifecycle monitoring, retraining governance, and explainability. The increasing use of cloud-based platforms, autonomous controls, and data-driven decision-making further necessitates continuous oversight and performance verification, particularly in Good Manufacturing Practice (GMP) environments.
While risk-based validation remains foundational, it too must expand to accommodate novel risk vectors introduced by AI such as algorithmic bias, data drift, adversarial vulnerability, and lack of interpretability. Existing standards like GAMP 5 and ICH Q9(R1) provide scaffolding for risk categorization, but AI systems require enhanced tools for data governance, bias auditing, and model transparency. Industry efforts must incorporate AI-specific risk matrices and audit trails that document decision logic, model lineage, and validation evidence throughout the system’s lifecycle.
To enable safe, reliable, and ethical use of AI in the pharmaceutical domain, regulatory frameworks must undergo systematic modernization. The FDA’s recent draft guidance on AI/ML-enabled medical devices, EMA’s AI reflection papers, and ISO/IEC 42001 signal a recognition of this need. However, guidance across agencies remains fragmented, and many national regulatory bodies lack the infrastructure or expertise to review AI-based submissions rigorously. Harmonized international frameworks and regulatory capacity-building initiatives will be essential to avoid compliance gaps and ensure global consistency.
Finally, national and global alignment is essential to ensure both product quality and patient safety. AI-enabled systems often operate across borders sourcing data from one region, processing in another, and making decisions that affect patients globally. Regulatory cooperation through bodies like ICH, PIC/S, and emerging FDA-EMA joint task forces can help facilitate common standards, mutual recognition, and faster adoption of best practices. Such collaboration is especially important for enabling AI in clinical development, pharmacovigilance, and post-marketing surveillance, where transnational data sharing and algorithm validation are essential.
In conclusion, the validation of AI/ML systems in pharmaceutical environments is both a scientific challenge and a regulatory imperative. To realize the promise of intelligent automation while preserving the principles of product integrity and patient safety, stakeholders must embrace flexible, transparent, and harmonized validation strategies guided by evolving standards, real-world evidence, and a shared commitment to innovation and compliance.
Acknowledgement
Authors would like to express their sincere gratitude to Principal Dr. S. J. Kshirsagar, MET’s Institute of Pharmacy for providing necessary facilities, to Dr. P. Kshirsagar, AI/ML expert and graduate of the Tata Institute of Social Sciences, Mumbai, INDIA for her practical guidance and deep understanding of Artificial Intelligence and Machine Learning (AI/ML), which significantly contributed to the technical depth of this review. Also thankful to Mr. A. Kshirsagar, a public policy expert and graduate of the University of Chicago, USA for his valuable support in identifying policy perspectives and regulatory gaps critical to the responsible implementation of AI/ML in the pharmaceutical sector. Special appreciation is extended to A. Deshmukh, Project Manager at Piramal, Inc., USA for his insights into the real-world integration of AI/ML within pharmaceutical project management.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Authors Contributions
Manish Dyaneshwar Baviskar: Data Collection, writing and editing
Sandeep Suresh Sonawane: Analysis, writing, editing and review
References:
- International Society of Automation (ISA). ISA-88: batch control and ISA-95: enterprise-control system integration standards. Research Triangle Park, NC: ISA; 2022.
- Gupta A, Kumar N, Shukla R. Artificial intelligence in GMP pharmaceutical manufacturing: challenges, regulatory guidelines, and future directions. J Pharm Innov. 2024;19(2):Epub ahead of print. Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC12195787. Accessed September 1, 2025.
- U.S. Food and Drug Administration (FDA). Using artificial intelligence & machine learning in the development of drug and biological products: discussion paper. Silver Spring, MD: FDA; May 2023.
- Babic B, Cohen IG, Stern AD, Li Y, Ouellet M. A general framework for governing marketed AI/ML medical devices via post-market surveillance. NPJ Digit Med. 2025;8(1):328. doi:10.1038/s41746-025-01717-9 PubMed
CrossRef - U.S. Food and Drug Administration (FDA). Marketing submission recommendations for AI-enabled device software functions: draft guidance for industry. Silver Spring, MD: FDA; January 6, 2025.
- U.S. Food and Drug Administration (FDA). Considerations for the use of artificial intelligence to support regulatory decision making for drug and biological products: draft guidance. Silver Spring, MD: FDA; January 7, 2025.
- International Society for Pharmaceutical Engineering (ISPE). GAMP® good practice guide: validation and lifecycle management of AI systems in regulated environments. Tampa, FL: ISPE; February 2025.
- European Commission. Artificial Intelligence Act – Regulation (EU) 2024/0130. Brussels: European Parliament and Council; 2024.
- European Medicines Agency (EMA). Reflection paper on the use of artificial intelligence in the medicinal product lifecycle. Amsterdam: EMA; December 2024.
- U.S. Food and Drug Administration (FDA). 21 CFR Part 11: electronic records; electronic signatures. Silver Spring, MD: FDA; 1997.
- European Medicines Agency (EMA). Annex 11 to the EU guidelines for good manufacturing practice: computerized systems. London: EMA; 2011.
- International Organization for Standardization (ISO). ISO/IEC 42001:2023 – artificial intelligence management system – requirements with guidance. Geneva: ISO; 2023.
- Medicines and Healthcare Products Regulatory Agency (MHRA). GXP data integrity guidance and definitions. London: MHRA; March 2022.
- CEN-CENELEC. CWA 18211:2025 – reference architecture for trustworthy AI in regulated industrial applications. Brussels: CEN-CENELEC; 2025.
- Niazi SK. Lifecycle validation of AI/ML systems in GMP environments: a regulatory perspective. J Pharm Sci Technol. 2025;79(3):233–242.
- International Organization for Standardization (ISO). ISO/IEC 27001:2022 – information security, cybersecurity and privacy protection – information security management systems – requirements. Geneva: ISO; 2022.
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Q8(R2) Pharmaceutical Development
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Q9(R1) Quality Risk Management
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Q10 Pharmaceutical Quality System; ICH Q11 Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities).
- International Organization for Standardization (ISO). ISO/IEC TR 24029-1:2021 – Artificial Intelligence (AI) — Assessment of the Robustness of Neural Networks — Part 1: Overview. Geneva: ISO; 2021.
Abbreviations List
AI – Artificial Intelligence.
ML – Machine Learning
GAMP – Good Automated Manufacturing Practice
ALCOA++ – Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available
ICH – International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
FDA – U.S. Food and Drug Administration
aMD – Software as a Medical Device
EU – European Union
ISO/IEC – International Organization for Standardization / International Electrotechnical Commission
IQ – Installation Qualification
OQ – Operational Qualification
PQ – Performance Qualification
GxP – Good Practice (covering GMP, GCP, GLP, etc.)
GMP – Good Manufacturing Practice
EMA – European Medicines Agency
ISPE – International Society for Pharmaceutical Engineering
COU – Context of Use
HMI – Human-Machine Interface
PLC – Programmable Logic Controller
SCADA – Supervisory Control and Data Acquisition
MES – Manufacturing Execution System
DCS – Distributed Control System
QMS – Quality Management System
PCCP – Predetermined Change Control Plan
SHAP – SHapley Additive explanations
LIME – Local Interpretable Model-Agnostic Explanations
XAI – Explainable Artificial Intelligence
KPIs – Key Performance Indicators
OECD – Organisation for Economic Co-operation and Development
WHO – World Health Organization
CEN-CENELEC – European Committee for Standardization / European Committee for Electrotechnical Standardization
ISA – International Society of Automation
Accepted on: 11-09-2025
Second Review by: Dr. Ana Golez
Final Approval by: Dr. Eugene A. Silow








