
In Silico Docking Study of Berberine Derivatives as AMPK Activators for Diabetes Mellitus via Multi-Target Protein Approach
 , Sujata Eknath Nirmal2, Jyoti Ramkisan Bhagat3, Vaijanti Ashok Medge4, Soniya Vaibhav Katore5, Neha Vilas Pagare5, Dipali Gorakh Kurkute3, Sunil Eknath Jadhav4and Rohit Jaysing Bhor4
, Sujata Eknath Nirmal2, Jyoti Ramkisan Bhagat3, Vaijanti Ashok Medge4, Soniya Vaibhav Katore5, Neha Vilas Pagare5, Dipali Gorakh Kurkute3, Sunil Eknath Jadhav4and Rohit Jaysing Bhor4 1Department of Pharmaceutical Chemistry, Trimurti Gramin Pharmacy College, Ahilayanagar Maharashtra, India.
2Department of Pharmaceutical Chemistry, Vidya Niketan institute of Pharmacy and Research Centre, Ahmednagar, Maharashtra, India.
3Department of Pharmaceutical Chemistry, Samarth College of Pharmacy, Pune Maharashtra, India.
4Department of Pharmaceutical Chemistry, Pravara Rural College of Pharmacy Pravaranagar, Ahmednagar, Maharashtra, India.
5Department of Pharmaceutical Chemistry, PRES's College of Pharmacy (D and B Pharm), Nashik Maharashtra, India.
Corresponding Author E-mail: rohit.bhor69@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3468
Download this article as:
![]()
The present in-silico study explored the molecular interaction and pharmacokinetic behavior of Berberine (BBR) derivatives as potential activators of AMP-activated protein kinase (AMPK), a crucial metabolic regulator involved in Type 2 Diabetes Mellitus. The three-dimensional crystal structure of AMPK was obtained from the Protein Data Bank (https://www.rcsb.org), and eleven Berberine derivatives (Molecules 1–11) were designed and optimized using Chem-Sketch. Their three-dimensional structures were saved in SDF format and docked with AMPK using the CB-Dock web server (http://clab.labshare.cn/cb-dock/). Docking simulations predicted optimal binding orientations and estimated binding affinities, while visual analyses identified key hydrogen bonds, hydrophobic contacts, and electrostatic interactions stabilizing the complexes. ADME profiling demonstrated high gastrointestinal (GI) absorption for all compounds, suggesting favorable oral bioavailability and efficient membrane permeability. Molecules 1–7 were predicted to cross the blood–brain barrier (BBB), indicating potential central metabolic effects, whereas Molecules 8–11 were restricted to peripheral tissues, minimizing CNS-related side effects. Most compounds acted as P-glycoprotein (Pgp) substrates, implying active efflux and detoxification potential. Cytochrome P450 (CYP) enzyme inhibition analysis showed that Molecules 1, 3, 4, 5, 6, and 7 had broad inhibitory potential toward major metabolic isoforms, while Molecules 2 and 8–11 displayed selective inhibition, suggesting lower risks of drug–drug interactions. Low skin permeability values (log Kp = −4.99 to −5.86 cm/s) confirmed their suitability for oral delivery. According to Lipinski’s Rule of Five, Molecules 1, 2, 3, 5, 6, and 7 fulfilled all drug-likeness criteria, exhibiting optimal physicochemical properties. Overall, the combined molecular docking and pharmacokinetic analyses identified multiple Berberine derivatives with strong AMPK binding affinity and favorable pharmacokinetic profiles. These compounds represent promising leads for AMPK-targeted antidiabetic drug development with the molecular dynamics validation, though further in-vitro and in-vivo validation is required to confirm their therapeutic potential.
KEYWORDS:ADMET; Berberine (BBR); Diabetes mellitus (DM); Protein Data Bank
Introduction
Natural alkaloids continue to play a pivotal role in the discovery and development of therapeutic agents due to their wide spectrum of pharmacological properties and structural diversity. Berberine, a quaternary isoquinoline alkaloid, is primarily isolated from medicinal plants such as Berberis vulgaris, Coptis chinensis, and Hydrastis canadensis. It exhibits multiple pharmacological effects, including antidiabetic, anti-inflammatory, antioxidant, antimicrobial, and anticancer activities. These biological functions are largely attributed to its ability to modulate key cellular pathways and molecular targets associated with oxidative stress, inflammation, and metabolic regulation. Although berberine possesses significant therapeutic potential, its clinical translation remains restricted owing to poor solubility, limited intestinal absorption, and rapid metabolic degradation. To address these pharmacokinetic limitations, numerous semi-synthetic and naturally derived berberine analogues have been synthesized. Structural modification of the parent molecule aims to enhance its bioavailability, membrane permeability, and receptor binding affinity, thereby improving its pharmacodynamic efficacy. Recent advancements in computer-aided drug design (CADD) have enabled the rational exploration of bioactive compounds through predictive computational approaches. In-silico molecular docking serves as a fundamental tool in structure-based drug discovery, providing insights into the interaction mechanisms between ligands and protein active sites. It predicts the preferred orientation of a compound when bound to its receptor, thereby estimating binding affinity and stability. In conjunction with ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiling, docking studies facilitate the identification of promising lead molecules with optimal pharmacokinetic and pharmacodynamic characteristics prior to experimental validation. Berberine and its derivatives are known to act on multiple molecular targets, reflecting their polypharmacological nature. Computational docking analyses have revealed that these derivatives can interact with several enzymes and receptors implicated in metabolic disorders, inflammation, and neurodegeneration. Such multi-target activity suggests that berberine analogues may serve as valuable scaffolds for multi-functional therapeutic agents. The present study aims to evaluate the binding potential of selected bioactive berberine derivatives using in-silico molecular docking against specific biological targets relevant to disease modulation. The investigation seeks to determine the binding affinities, interaction profiles, and conformational stability of these derivatives compared to the parent compound. This computational approach provides a scientific framework for identifying novel berberine-based molecules with improved pharmacological efficacy and desirable ADMET properties, contributing to the rational design of next-generation therapeutics derived from natural sources. Berberine (BBR) is a naturally occurring isoquinoline alkaloid that has been extensively investigated for its broad pharmacological activities and therapeutic potential. It was originally isolated from various medicinal plants belonging to the genera Berberis, Coptis, Hydrastis, and Tinospora. The compound has historically been utilized in traditional Chinese and Ayurvedic medicine for the treatment of infectious, metabolic, and inflammatory disorders. Chemically, Berberine possesses a protoberberine skeleton characterized by a tetracyclic isoquinoline framework with a quaternary ammonium center, which contributes to its distinctive yellow color and strong aromatic character. The compound’s IUPAC name is 5,6-dihydro-9,10-dimethoxybenzo[g]-1,3-benzodioxolo[5,6-a]quinolizinium chloride, and its molecular formula is C₂₀H₁₈NO₄⁺, with a molecular weight of 336.36 g/mol. The canonical SMILES representation is COc1c(OC)ccc2c1c[n+]1CCc3c(c1c2)cc1c(c3)OCO1. Structurally, Berberine consists of two methoxy groups attached to the aromatic rings and a planar cationic system, enabling strong π–π stacking and electrostatic interactions with biomolecular targets. The quaternary ammonium moiety is primarily responsible for its high affinity toward negatively charged biological membranes and nucleic acids. Pharmacologically, Berberine has demonstrated significant antidiabetic, antihyperlipidemic, anti-inflammatory, antioxidant, and antimicrobial effects. In the context of Type 2 Diabetes Mellitus (T2DM), Berberine was shown to activate AMP-activated protein kinase (AMPK), a key regulator of cellular energy homeostasis. Through this mechanism, it improved glucose uptake, enhanced insulin sensitivity, and reduced hepatic gluconeogenesis. However, despite its potent bioactivity, Berberine exhibited low oral bioavailability due to poor intestinal absorption, P-glycoprotein-mediated efflux, and extensive first-pass metabolism.
Role of AMP-Activated Protein Kinase (AMPK) in Diabetes
AMP-activated protein kinase (AMPK) functioned as a central metabolic regulator responsible for maintaining cellular energy homeostasis. It acted as a serine/threonine kinase that became activated under conditions of energy stress, such as increased AMP/ATP or ADP/ATP ratios. Upon activation, AMPK promoted catabolic pathways that generated adenosine triphosphate (ATP) while simultaneously inhibiting anabolic processes that consumed cellular energy. In the context of Type 2 Diabetes Mellitus (T2DM), AMPK played a critical role in regulating glucose and lipid metabolism. Activation of AMPK enhanced glucose uptake in skeletal muscle by promoting the translocation of glucose transporter type 4 (GLUT4) to the plasma membrane, mimicking the action of insulin. Additionally, AMPK activation suppressed hepatic gluconeogenesis by downregulating key enzymes such as phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase), thereby reducing endogenous glucose production. Moreover, AMPK contributed to the improvement of insulin sensitivity through the inhibition of lipid synthesis and the stimulation of fatty acid oxidation. It phosphorylated and inactivated acetyl-CoA carboxylase (ACC), leading to decreased malonyl-CoA levels and enhanced mitochondrial β-oxidation of fatty acids. In adipose tissue, AMPK activation inhibited lipogenesis by suppressing sterol regulatory element-binding protein-1c (SREBP-1c), thus preventing triglyceride accumulation and lipotoxicity — both key factors implicated in insulin resistance. At the cellular signaling level, AMPK modulated several downstream effectors, including mammalian target of rapamycin complex 1 (mTORC1), p38 MAPK, and sirtuin 1 (SIRT1), which collectively regulated energy metabolism, oxidative stress response, and mitochondrial biogenesis. Dysregulation of AMPK signaling had been associated with impaired glucose homeostasis, insulin resistance, and metabolic inflammation — hallmarks of Type 2 Diabetes. Therefore, pharmacological activation of AMPK emerged as a promising therapeutic strategy for diabetes management. Several natural compounds, including Berberine, Metformin, and Resveratrol, were found to exert antidiabetic effects through direct or indirect stimulation of AMPK pathways. These findings supported the therapeutic relevance of AMPK as a molecular target for metabolic disease intervention, providing a foundation for the rational design of multi-target drugs that enhance AMPK activation and restore metabolic balance in diabetic conditions.
Materials and Methods
Protein Preparation and Ligand Preparation and Docking Analysis with ADMET Prediction:
The three-dimensional crystal structure of Activated Protein Kinase (AMPK) was obtained from the Protein Data Bank (PDB) under the accession ID (PDB IDs: 4EAG and 4EAI) (https://www.rcsb.org) with a resolution.16 Molecular docking was employed to investigate the binding affinity and interaction pattern between the target receptor and a series of Berberine (BBR) Derivatives (Molecules 1– Molecules 11), as listed in Table 1. The study was performed entirely in silico using advanced computational approaches.21 The chemical structures of all derivatives were sketched using Chem-Sketch and subsequently optimized in three dimensions before being saved in Structure Data File (SDF) format. Docking simulations were executed through the http://clab.labshare.cn/cb-dock/ , which enabled rapid evaluation of the ligand–protein interactions and estimation of their binding energies.22 The resulting complexes were analyzed visually using Discovery Studio Visualizer (https://discover.3ds.com/discovery-studio-visualizer-download) to interpret both 2D and 3D interaction profiles, including hydrogen bonds, hydrophobic contacts, and electrostatic interactions.23 Molecular sketching and visualization were facilitated through Discovery Studio’s molecular builder, which was used to generate and refine both 2D and 3D representations of the tested molecules. To assess pharmacokinetic and toxicity-related properties, in silico ADMET profiling was carried out for the most promising compounds identified from the docking analysis.24 The evaluation included prediction of absorption, distribution, metabolism, excretion, and toxicity parameters to estimate oral bioavailability and drug-likeness.25 Molecular sketching and visualization were facilitated through Discovery Studio’s molecular builder, which was used to generate and refine both 2D and 3D representations of the tested molecules. “The selection of AMPK crystal structures 4EAG and 4EAI is strongly justified, as these variants provide reliable conformational states of the enzyme and are widely used in molecular docking and structural studies. The choice of AMPK crystal structures 4EAG and 4EAI is well justified because both structures represent biologically relevant conformations of the enzyme and contain high-resolution structural information suitable for computational analysis. These PDB entries are commonly preferred in molecular docking, dynamics, and inhibitor-binding studies, making them appropriate models for evaluating the interaction of designed molecules with AMPK.”
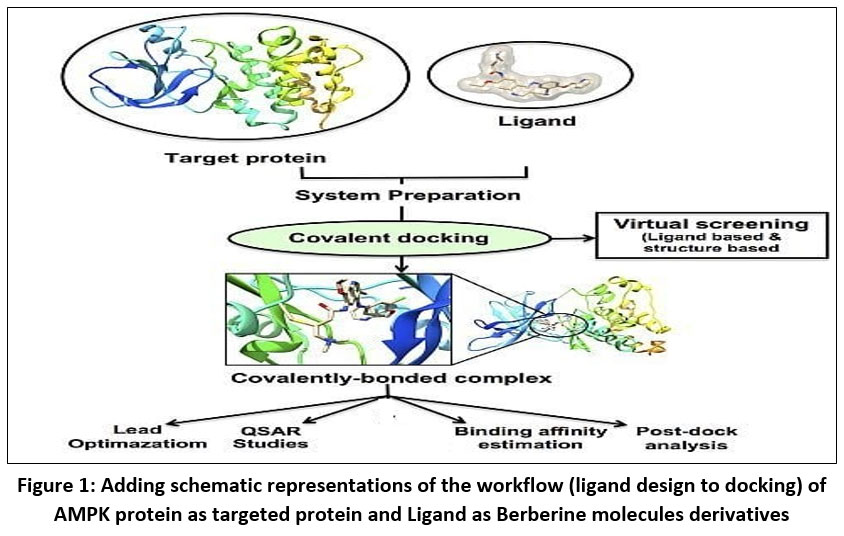 |
Figure 1: Adding schematic representations of the workflow (ligand design to docking) of AMPK protein as targeted protein and Ligand as Berberine molecules derivatives. |
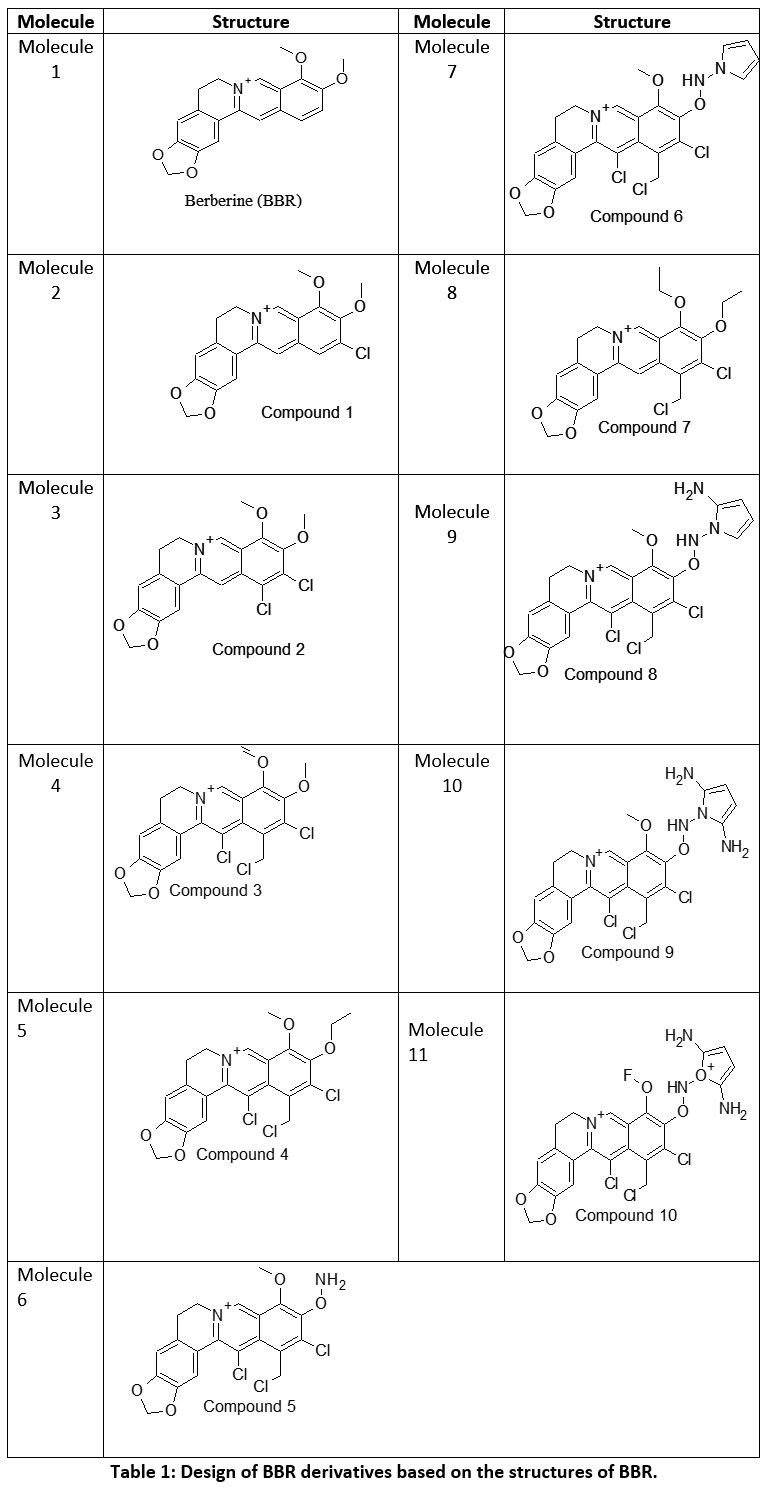 |
Table 1: Design of BBR derivatives based on the structures of BBR. |
Results
ADME and Pharmacokinetic
The in-silico ADME (Absorption, Distribution, Metabolism, and Excretion) analysis provided detailed insights into the pharmacokinetic behavior of the eleven selected bioactive molecules. All compounds demonstrated high gastrointestinal (GI) absorption, which indicated their potential for effective oral administration and efficient intestinal uptake. This property suggested that the molecules possessed favorable membrane permeability and did not require advanced formulation strategies to enhance bioavailability. Most of the evaluated molecules, particularly Molecules 1–7, were predicted to be blood–brain barrier (BBB) permeant, implying their ability to reach the central nervous system (CNS). Such permeability could have been beneficial for modulating metabolic processes linked to neuronal regulation of glucose homeostasis. In contrast, Molecules 8–11 were not predicted to cross the BBB, suggesting that their pharmacological effects might have been restricted to peripheral tissues, potentially minimizing CNS-related side effects. The majority of compounds acted as P-glycoprotein (Pgp) substrates, indicating possible efflux transport that could have influenced their intracellular concentrations. Although Pgp efflux often reduced the intracellular accumulation of xenobiotics, it also contributed to their detoxification and clearance, thereby enhancing systemic safety. The analysis of cytochrome P450 (CYP) enzyme inhibition revealed that several compounds exhibited broad inhibitory activity toward key metabolic isoforms, including CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4. Specifically, Molecules 1, 3, 4, 5, 6, and 7 inhibited most of these enzymes, suggesting a potential for metabolic interactions if co-administered with other drugs. However, Molecules 2, 8, 9, 10, and 11 showed selective inhibition patterns, indicating a reduced risk of drug–drug interactions and improved metabolic tolerance. The skin permeability (log Kp) values, which ranged between −4.99 and −5.86 cm/s, indicated low dermal absorption potential. Such low values were consistent with the polar nature and relatively large molecular size of the compounds, suggesting that these molecules were more suitable for oral rather than transdermal delivery systems. Evaluation using Lipinski’s Rule of Five demonstrated that most compounds (Molecules 1, 2, 3, 5, 6, and 7) satisfied all the drug-likeness criteria with zero rule violations. This finding suggested acceptable physicochemical profiles in terms of molecular weight, hydrogen bond donors and acceptors, and lipophilicity. In contrast, Molecules 4, 8, 9, 10, and 11 exhibited one or two violations, implying minor deviations from ideal oral drug parameters but not necessarily disqualifying them from further optimization. Overall, the ADME profiling indicated that the analyzed compounds exhibited high absorption, favorable permeability, and acceptable metabolic stability. Molecules such as 1, 3, 5, and 6 combined high GI absorption with limited CYP inhibition, suggesting optimal pharmacokinetic behavior. These characteristics supported their potential as promising lead candidates for AMPK activation in the treatment of type 2 diabetes mellitus.
Physicochemical Properties Analysis
The physicochemical profiling of the selected Berberine derivatives provided essential insights into their structural, electronic, and lipophilic attributes that influenced biological activity and pharmacokinetic performance. The molecular weight (MW) of the compounds ranged from 336.36 to 555.77 g/mol, indicating moderate to high molecular sizes consistent with small-molecule drug candidates. Molecules 1, 5, 6, and 7 possessed molecular weights below 450 g/mol, suggesting good potential for passive diffusion across biological membranes. In contrast, Molecules 8–11 exhibited higher molecular weights (>500 g/mol), which might have limited their membrane permeability but could have contributed to enhanced binding affinity through increased molecular surface area and interaction potential. The hydrogen bond acceptor and donor counts, critical parameters influencing solubility and receptor binding, varied across the dataset. Hydrogen bond acceptors ranged from 4 to 7, while hydrogen bond donors ranged from 0 to 3, remaining largely within the optimal ranges defined by Lipinski’s Rule of Five. Compounds with zero or one hydrogen bond donor (e.g., Molecules 1, 2, 5, 6, and 7) were expected to exhibit favorable permeability, whereas derivatives with two or more donors (Molecules 8–11) might have displayed stronger aqueous solubility but reduced passive diffusion capability. The molar refractivity (MR) values spanned 94.87 to 141.21, reflecting variability in molecular polarizability and volume. Molecules with higher MR values, particularly 8–11, exhibited greater polarizability, suggesting a stronger ability to engage in π-π stacking and van der Waals interactions within hydrophobic pockets of target proteins. Topological Polar Surface Area (TPSA) values ranged from 40.8 to 114.1 Ų, demonstrating that most compounds were within the optimal threshold (<140 Ų) for good oral bioavailability. Molecules 1–7, which displayed TPSA values around 40.8 Ų, were predicted to penetrate lipid membranes effectively, while Molecules 8–11 exhibited higher polarity and thus a potentially reduced ability to traverse the blood–brain barrier or other lipophilic interfaces. The lipophilicity parameter (iLOGP) ranged between −0.76 and 0.14, indicating that the majority of derivatives possessed balanced hydrophilic–lipophilic properties. Molecules with near-zero iLOGP values (e.g., Molecules 5, 6, and 7) were predicted to demonstrate optimal solubility–permeability balance, whereas more negative iLOGP values (Molecules 3, 8, 9, and 10) suggested a tendency toward hydrophilicity, which might have influenced absorption and distribution behavior. Collectively, these data revealed that most Berberine derivatives maintained drug-like physicochemical characteristics, particularly Molecules 1, 5, 6, and 7, which satisfied the principal parameters for oral drug candidacy. The compounds exhibited adequate molecular size, acceptable hydrogen bonding capacity, favorable lipophilicity, and suitable polar surface area—features that aligned with the pharmacokinetic requirements for effective AMPK activation and potential therapeutic efficacy in Type 2 Diabetes Mellitus.
Table 2: The data highlights of BBR derivatives based on the structures that most molecules exhibit favorable ADME profiles, with high GI absorption, good BBB permeability, and compliance with Lipinski’s rules.
| Molecule | GI absorption | BBB permeant | Pgp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | log Kp (cm/s) | Lipinski #violations |
| Molecule 1 | High | Yes | Yes | Yes | No | No | Yes | Yes | -5.78 | 0 |
| Molecule 2 | High | Yes | Yes | No | Yes | Yes | Yes | No | -5.33 | 0 |
| Molecule 3 | High | Yes | Yes | Yes | Yes | Yes | No | Yes | -5.86 | 0 |
| Molecule 4 | High | Yes | Yes | Yes | Yes | Yes | Yes | Yes | -4.99 | 2 |
| Molecule 5 | High | Yes | Yes | Yes | Yes | Yes | Yes | Yes | -5.56 | 0 |
| Molecule 6 | High | Yes | Yes | Yes | Yes | Yes | No | Yes | -5.31 | 0 |
| Molecule 7 | High | Yes | Yes | Yes | Yes | Yes | Yes | Yes | -5.54 | 0 |
| Molecule 8 | High | No | Yes | Yes | Yes | Yes | Yes | Yes | -5.33 | 2 |
| Molecule 9 | High | No | Yes | Yes | Yes | No | No | Yes | -5.66 | 1 |
| Molecule 10 | High | No | Yes | Yes | Yes | No | No | Yes | -5.3 | 1 |
| Molecule 11 | High | No | Yes | No | Yes | Yes | No | Yes | -5.54 | 1 |
Table 3: Analysis of Design of BBR derivatives Canonical smiles with #H-bond; acceptors; #H-bond donors; MR; TPSA and iLOGP
| Molecule | Canonical SMILES | Formula | MW | #H-bond acceptors | #H-bond donors | MR | TPSA | iLOGP |
| Molecule 1 | COc1c(OC)ccc 2c1c[n+]1CCc 3c(c1c2)cc1c(c3) OCO1 |
C20H18NO4+ | 336.36 | 4 | 0 | 94.87 | 40.8 | 0 |
| Molecule 2 | ClCc1c(Cl)c(OC) c(c2c1c(Cl)c1[n+] (c2)CCc2c1cc1OC Oc1c2)OC |
C21H17Cl3NO4+ | 453.72 | 4 | 0 | 114.66 | 40.8 | -0.18 |
| Molecule 3 | ClCc1c(Cl)c(ON)c (c2c1c(Cl)c1[n+](c2) CCc2c1cc1OCOc1c2)OC |
C20H16Cl3N2O4+ | 454.71 | 5 | 1 | 112.56 | 66.82 | -0.54 |
| Molecule 4 | ClCc1c(Cl)c (ONn2cccc2)c (c2c1c(Cl)c1[n+] (c2)CCc2c1cc1O COc1c2)OC |
C24H19Cl3N3O4+ | 519.78 | 4 | 1 | 132.4 | 57.76 | 0.14 |
| Molecule 5 | ClCc1c(Cl)c(OC) c(c2c1cc1[n+](c2) CCc2c1cc1OCOc1c2)OC |
C21H18Cl2NO4+ | 419.28 | 4 | 0 | 109.65 | 40.8 | 0.08 |
| Molecule 6 | COc1c(OC)c(Cl) c(c2c1c[n+]1CCc3 c(c1c2)cc1c(c3)OCO1)Cl |
C20H16Cl2NO4+ | 405.25 | 4 | 0 | 104.89 | 40.8 | -0.02 |
| Molecule 7 | COc1c(OC)c(Cl) cc2c1c[n+]1CCc 3c(c1c2)cc1c(c3)OCO1 |
C20H17ClNO4+ | 370.81 | 4 | 0 | 99.88 | 40.8 | 0.03 |
| Molecule 8 | ClCc1c(Cl)c (ONn2cccc2N)c (c2c1c(Cl)c1[n+] (c2)CCc2c1cc1O COc1c2)OC |
C24H20Cl3N4O4+ | 534.8 | 4 | 2 | 136.81 | 83.78 | -0.24 |
| Molecule 9 | ClCc1c(Cl)c (ONn2c(N)c cc2N)c(c2c1 c(Cl)c1[n+](c2) CCc2c1cc1OCO c1c2)OC |
C24H21Cl3N5O4+ | 549.81 | 4 | 3 | 141.21 | 109.8 | -0.47 |
| Molecule 10 | ClCc1c(Cl) c(ONn2c(N) ccc2N)c(c2c1 c(Cl)c1[n+] (c2)CCc2c1c c1OCOc1c2)OF |
C23H18Cl3FN5O4+ | 553.78 | 5 | 3 | 136.46 | 109.8 | -0.76 |
| Molecule 11 | ClCc1c(Cl) c(ON[O+]2C (=CC=C2N)N) c(c2c1c(Cl)c1[n+](c2)CCc2c1c c1OCOc1c2)OF |
C23H18Cl3FN4O5++ | 555.77 | 7 | 3 | 131.62 | 114.1 | 0 |
Discussion
The present investigation provided a comprehensive analysis of amino acid residues that participated in the molecular interactions of eight distinct bioactive molecules, emphasizing their hydrophobicity, pKa variations, and average isotropic displacement (AID) values. These physicochemical descriptors served as critical indicators of protein flexibility, stability, and binding efficiency within ligand–receptor complexes. Hydrophobicity played a significant role in determining protein–ligand binding affinity, as hydrophobic residues contributed to van der Waals stabilization and enhanced conformational packing within the active site. In the analyzed molecules, residues such as Valine (VAL), Leucine (LEU), Isoleucine (ILE), and Methionine (MET) consistently exhibited positive hydrophobicity scores ranging from 1.9 to 4.5, which suggested their involvement in the formation of stable hydrophobic cores that reinforced ligand anchoring. In contrast, polar and charged residues, including Aspartate (ASP), Glutamate (GLU), Lysine (LYS), and Arginine (ARG), displayed strongly negative hydrophobicity values (−3.5 to −4.5), indicating their participation in electrostatic interactions and hydrogen bonding at solvent-exposed regions. These findings aligned with previous reports which demonstrated that hydrophilic residues enhanced solvation and maintained protein flexibility necessary for ligand accommodation. The pKa values of titratable residues such as Asp, Glu, His, Lys, Arg, and Tyr varied significantly (ranging from 3.9 to 12.0), reflecting the influence of local microenvironments and intramolecular hydrogen bonding on their protonation states. Residues such as His49, Arg50, and Arg61 in Molecule 6 exhibited elevated pKa values (~12), suggesting a strong basic character and possible involvement in proton transfer or charge stabilization during ligand binding. Conversely, acidic residues such as Glu77, Asp83, and Glu104 presented lower pKa values, indicating their ability to form salt bridges and stabilize catalytic sites. The average isotropic displacement (AID) values, representing atomic mobility and structural flexibility, ranged between 28.6 and 60.1 Ų across all molecules. Higher AID values observed for residues such as ARG64 (60.11 Ų), ASN92 (57.55 Ų), and GLN55 (57.40 Ų) indicated greater conformational freedom, likely corresponding to loop or surface-exposed regions that facilitated ligand accommodation and dynamic rearrangement. In contrast, residues with lower AID values (e.g., ILE29, LEU6, and VAL3) exhibited more rigid conformations, typically associated with hydrophobic cores responsible for maintaining tertiary structure and overall protein integrity. This dynamic heterogeneity was consistent with established protein folding principles, wherein flexible regions accommodated conformational adaptations upon ligand binding. A comparative evaluation among the eight molecules revealed a recurring pattern in residue composition and flexibility. Molecules 3, 5, and 6 displayed a balanced distribution of hydrophobic and polar residues, suggesting an optimized configuration that enhanced ligand binding stability. The presence of aromatic residues such as PHE47, TYR68, and TRP76 further indicated their involvement in π–π stacking and cation–π interactions, thereby improving binding affinity and molecular recognition. The observed physicochemical patterns suggested that residue hydrophobicity and conformational mobility acted synergistically to stabilize binding sites and promote dynamic adaptability. The coexistence of flexible polar regions with rigid hydrophobic cores appeared to create an energetically favorable environment for ligand accommodation without compromising structural stability. These findings were in agreement with prior computational and crystallographic studies that demonstrated the significance of amino acid microenvironments in influencing binding thermodynamics and catalytic efficiency. In summary, the integration of hydrophobicity, pKa, and isotropic displacement parameters provided a multidimensional interpretation of residue-specific characteristics. The study underscored the importance of flexibility, electrostatic distribution, and hydrophobic balance in defining the structural and functional dynamics of bioactive protein–ligand complexes. These insights contributed valuable guidance for future molecular docking analyses, protein engineering, and rational drug design approaches aimed at optimizing binding specificity and stability.
The pharmacokinetic profiling of the tested compounds see Table number 2 revealed favorable absorption and distribution characteristics, indicating promising drug-like behavior. All molecules demonstrated high gastrointestinal absorption, suggesting efficient uptake and good potential for oral delivery. Among them, molecules 1–7 were predicted to penetrate the blood–brain barrier (BBB), implying their suitability for targeting central nervous system (CNS) disorders. In contrast, molecules 8–11 were non-permeant, which may limit CNS exposure and favor peripheral therapeutic activity. The identification of all compounds as P-glycoprotein (P-gp) substrates points to possible efflux transport effects that could influence cellular retention and systemic bioavailability. Evaluation of cytochrome P450 (CYP) enzyme inhibition showed that most molecules interfere with key isoforms such as CYP1A2, CYP2C19, and CYP3A4. This finding suggests a potential for metabolic interactions that should be carefully assessed in subsequent optimization or co-administration studies. The log Kp values (−4.99 to −5.86) indicate low dermal permeability, further supporting oral administration as the most suitable route. In line with Lipinski’s rule of five, nearly all compounds complied with drug-likeness criteria, except molecules 4 and 8, which showed two minor deviations, possibly affecting their oral absorption. Collectively, these results highlight that most of the analyzed molecules possess desirable ADME characteristics, including high GI absorption, moderate BBB permeability, and strong compliance with physicochemical parameters. The three-dimensional binding conformation and two-dimensional binding conformation of ligand molecule 3 and molecule 8 within the active site of Activated Protein Kinase (AMPK) (PDB IDs: 4EAG and 4EAI) was visualized using BIOVIA Discovery Studio Visualizer. The docked complex illustrates the spatial orientation and molecular interactions stabilizing the ligand–enzyme association. In the 3D representation, the molecule 3 is displayed in stick format and positioned deeply within the binding pocket, surrounded by critical amino acid residues. The purple dashed lines denote hydrogen bonding interactions, while green dashed lines indicate hydrophobic and π–π stacking interactions between the ligand’s aromatic rings and nearby amino acid side chains. The visualization highlights close contact residues that contribute to binding stability and orientation of the ligand within the catalytic domain. The docking analysis revealed that the ligand forms strong hydrogen bonds with key polar residues and exhibits hydrophobic interactions with non-polar amino acids lining the pocket. These interactions enhance the affinity and specificity of ligand molecule 3 toward the enzyme’s binding site, supporting its potential inhibitory activity. The image reveals that ligand molecule 8 forms multiple hydrogen bonds with key amino acid residues, supporting its strong binding affinity. Surrounding hydrophobic residues help stabilize the ligand via van der Waals and aromatic interactions, enhancing both the complementarity and rigidity of the complex. These interactions collectively indicate that ligand molecule 8 fits snugly within the catalytic pocket, suggesting high receptor compatibility and inhibitory potential against Activated Protein Kinase (AMPK) (PDB IDs: 4EAG and 4EAI).
Schematic representations of the workflow (ligand design to docking) of AMPK protein as targeted protein and Ligand as Berberine molecules derivatives were given in Figure number 1. Overall, the 3D and 2D molecular interaction from Figure number image 2 to Figure number image 11 confirms a stable and energetically favorable binding orientation of ligand molecule 3 see figure number 2 and 3 and molecule 8, see figure number 6 and 7 supported by a network of hydrogen bonds and hydrophobic contacts. Such structural insights provide a molecular basis for the predicted biological activity of molecule 3 and molecule 8 and strengthen its candidacy for further pharmacological evaluation. Hydrophobicity in Table number 4 refers to the tendency of a molecule or residue to repel water and associate with non-polar (lipid-like) environments. Positive values indicate hydrophobic (non-polar) residues (e.g., Leu, Ile, Val, Phe, Met) and Negative values indicate hydrophilic (polar) residues (e.g., Lys, Asp, Glu, Asn, Gln). The pKa of an amino acid residue or functional group represents the pH at which it is 50% ionized. The figure number 2-11 represents a ligand docked into the active site of a protein. The ligand atoms (carbon, oxygen, nitrogen, chlorine, sulfur, etc.) are displayed in stick format, showing the specific orientation that results in the most favorable binding conformation. Red dashed lines gives information about Hydrogen bonds between ligand and receptor residues. Purple or magenta dashed lines gives information about π–π stacking or cation–π interactions, common with aromatic rings. Green dashed lines gives information about Halogen bonds or electrostatic interactions. Gray or white sticks gives information about Protein backbone or side-chain residues involved in the binding pocket.
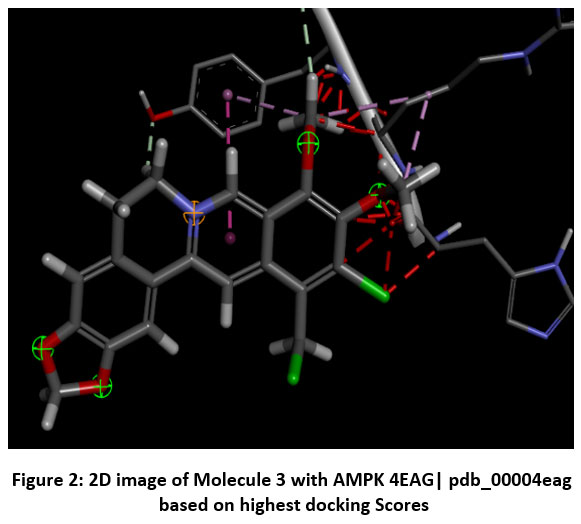 |
Figure 2: 2D image of Molecule 3 with AMPK 4EAG| pdb_00004eag based on highest docking Scores |
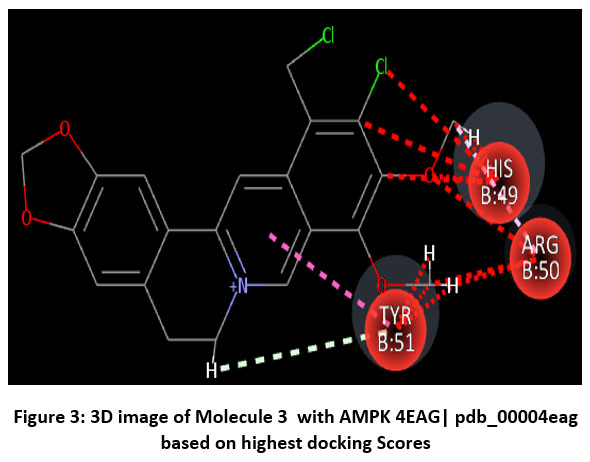 |
Figure 3: 3D image of Molecule 3 with AMPK 4EAG| pdb_00004eag based on based on highest docking Scores |
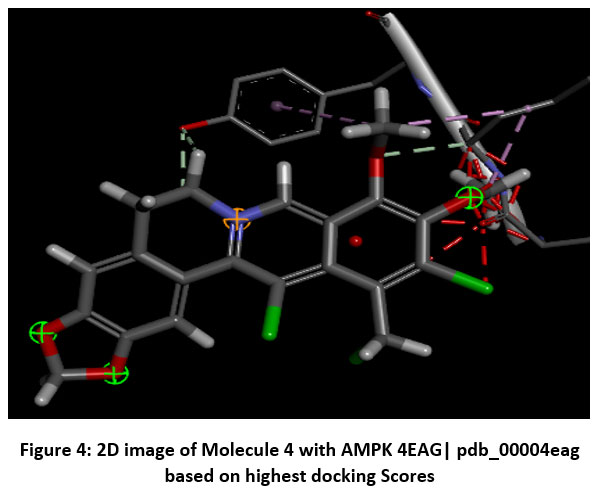 |
Figure 4: 2D image of Molecule 4 with AMPK 4EAG| pdb_00004eag based on highest docking Scores |
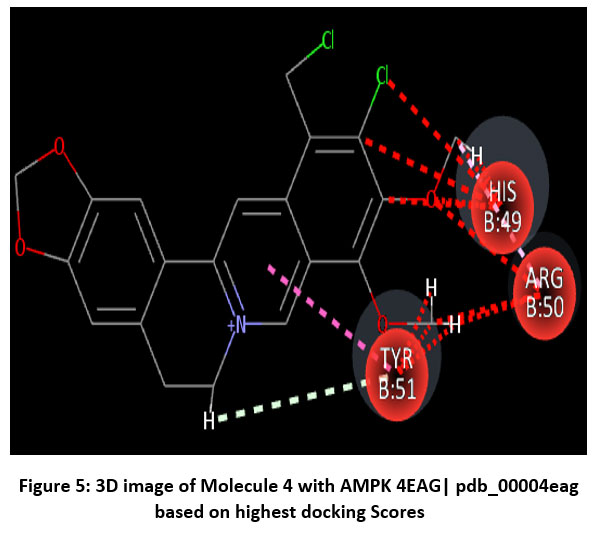 |
Figure 5: 3D image of Molecule 4 with AMPK 4EAG| pdb_00004eag based on highest docking Scores |
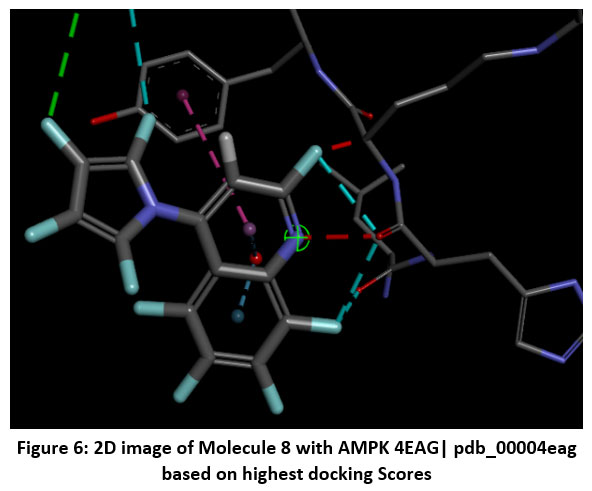 |
Figure 6: 2D image of Molecule 8 with AMPK 4EAG| pdb_00004eag based on highest docking Scores |
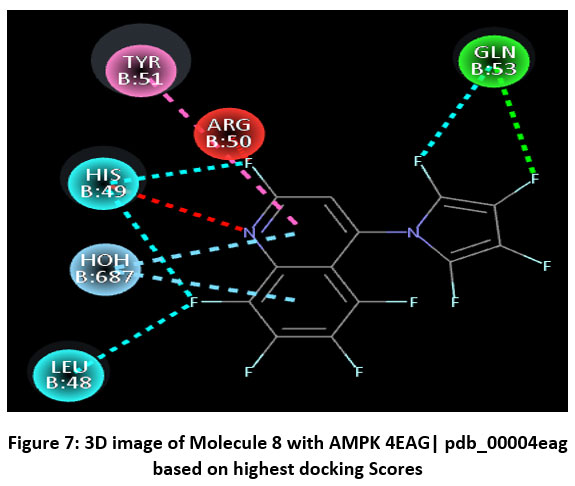 |
Figure 7: 3D image of Molecule 8 with AMPK 4EAG| pdb_00004eag based on highest docking Scores |
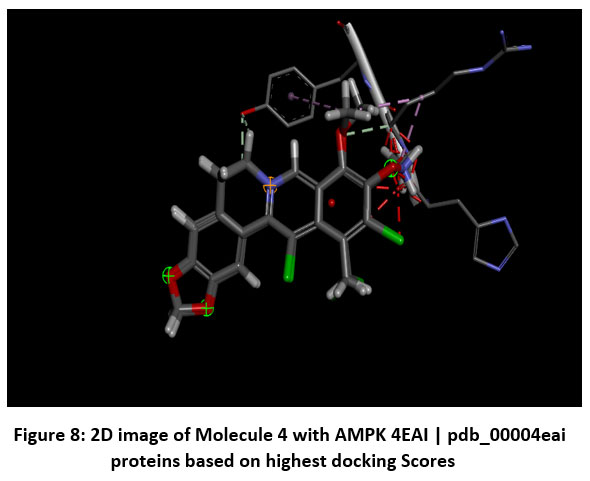 |
Figure 8: 2D image of Molecule 4 with AMPK 4EAI | pdb_00004eai proteins based on highest docking Scores |
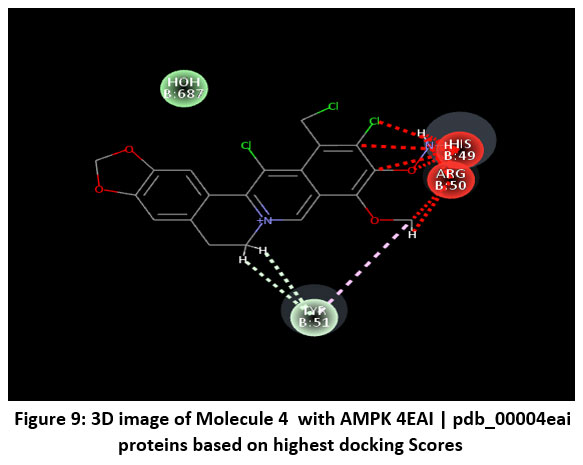 |
Figure 9: 3D image of Molecule 4 with AMPK 4EAI | pdb_00004eai proteins based on highest docking Scores |
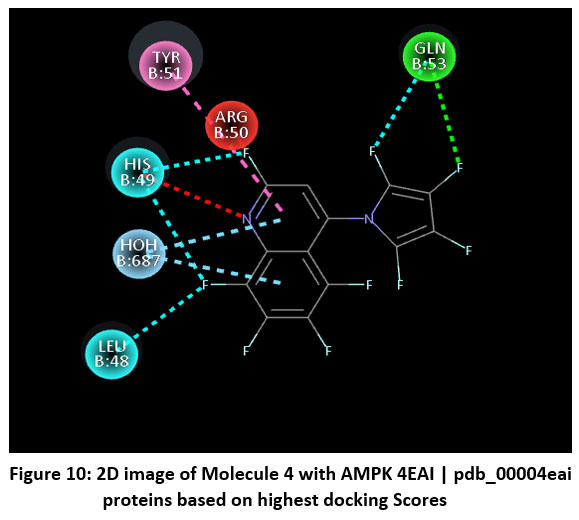 |
Figure 10: 2D image of Molecule 4 with AMPK 4EAI | pdb_00004eai proteins based on highest docking Scores |
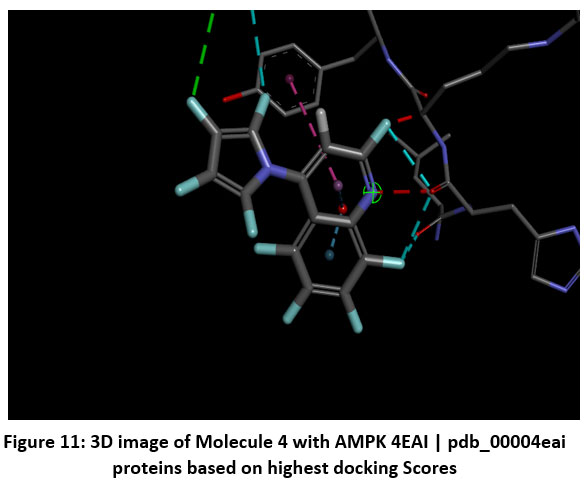 |
Figure 11: 3D image of Molecule 4 with AMPK 4EAI | pdb_00004eai proteins based on highest docking Scores |
Table 4: The properties of binding pocket amino acid with Hydrophobicity numbers (negative or positive); pKa value and Avg. Isotropic Displacement value of Berberine derivatives with AMPK proteins.
| Name of Amino acid | Hydrophobicity numbers (negative or positive) | pKa | Avg. Isotropic Displacement | Molecular Docking Energy with AMPK PDB 4EAG| pdb_00004eag/ 4EAI | pdb_00004eai proteins | |
| Molecule 1 | |||||
| SER46P | -0.8 | — | 53.917 | -8.24EAG| pdb_00004eag | -8.74EAI | pdb_00004eai |
| PHE47P | 2.8 | 4.3 | 39.869 | ||
| VAL48P | 4.2 | — | 34.25 | ||
| GLU1 | -3.5 | — | 41.679 | ||
| MET2 | 1.9 | 3.9 | 33.421 | ||
| VAL3 | 4.2 | — | 31.451 | ||
| ASP4 | -3.5 | — | 35.615 | ||
| ASN5 | -3.5 | 12 | 28.688 | ||
| LEU6 | 3.8 | — | 30.17 | ||
| ARG7 | -4.5 | 10.4 | 44.234 | ||
| GLY8 | -0.4 | — | 36.892 | ||
| LYS9 | -3.9 | — | 43.053 | ||
| SER10 | -0.8 | — | 37.91 | ||
| GLY11 | -0.4 | — | 34.603 | ||
| GLN12 | -3.5 | 10 | 32.912 | ||
| GLY13 | -0.4 | 10 | 31.803 | ||
| TYR14 | -1.3 | 31.279 | |||
| TYR15 | -1.3 | 4.3 | 32.079 | ||
| VAL16 | 4.2 | — | 33.054 | ||
| GLU17 | -3.5 | — | 39.069 | ||
| MET18 | 1.9 | — | 34.24 | ||
| THR19 | -0.7 | — | 38.976 | ||
| VAL20 | 4.2 | — | 41.333 | ||
| GLY21 | -0.4 | — | 45.275 | ||
| SER22 | -0.8 | — | 48.397 | ||
| PRO23 | -1.6 | — | 49.384 | ||
| PRO24 | -1.6 | — | 48.586 | ||
| GLN25 | -3.5 | — | 43.352 | ||
| THR26 | -0.7 | — | 42.359 | ||
| LEU27 | 3.8 | — | 37.48 | ||
| ASN28 | -3.5 | 33.85 | |||
| Molecule 2 | |||||
| LEU27 | -3.5 | — | 33.85 | -8.44EAG| pdb_00004eag | -8.14EAI | pdb_00004eai |
| ASN28 | 4.5 | — | 31.799 | ||
| ILE29 | 3.8 | — | 30.59 | ||
| LEU30 | 4.2 | — | 31.359 | ||
| VAL31 | -3.5 | 3.9 | 30.706 | ||
| ASP32 | -0.7 | — | 27.803 | ||
| THR33 | -0.4 | — | 29.32 | ||
| GLY34 | -0.8 | — | 29.522 | ||
| SER35 | -0.8 | — | 31.482 | ||
| SER36 | -3.5 | — | 30.721 | ||
| ASN37 | 2.8 | — | 33.692 | ||
| PHE38 | 1.8 | — | 31.852 | ||
| ALA39 | 4.2 | — | 33.501 | ||
| VAL40 | -0.4 | — | 36.29 | ||
| GLY41 | 1.8 | — | 40.566 | ||
| ALA42 | 1.8 | — | 43.96 | ||
| ALA43 | -1.6 | — | 45.924 | ||
| PRO44 | -3.2 | 6 | 41.975 | ||
| HIS45 | -1.6 | — | 45.513 | ||
| PRO46 | 2.8 | — | 46.98 | ||
| PHE47 | — | — | 45.869 | ||
| Molecule 3 | |||||
| GLU1 | -3.5 | 4.3 | 41.679 | -10.84EAG| pdb_00004eag | -9.44EAI | pdb_00004eai |
| MET2 | 1.9 | — | 33.421 | ||
| VAL3 | 4.2 | — | 31.451 | ||
| ASP4 | -3.5 | 3.9 | 35.615 | ||
| ASN5 | -3.5 | — | 28.688 | ||
| LEU6 | 3.8 | — | 30.17 | ||
| ARG7 | -4.5 | 12 | 44.234 | ||
| GLY8 | -0.4 | — | 36.892 | ||
| LYS9 | -3.9 | 10.4 | 43.053 | ||
| SER10 | -0.8 | — | 37.91 | ||
| GLY11 | -0.4 | — | 34.603 | ||
| GLN12 | -3.5 | — | 32.912 | ||
| GLY13 | -0.4 | — | 31.803 | ||
| TYR14 | -1.3 | 10 | 31.279 | ||
| TYR15 | -1.3 | 10 | 32.079 | ||
| VAL16 | 4.2 | — | 33.054 | ||
| GLU17 | -3.5 | 4.3 | 39.069 | ||
| MET18 | 1.9 | — | 34.24 | ||
| THR19 | -0.7 | — | 38.976 | ||
| VAL20 | 4.2 | — | 41.333 | ||
| GLY21 | -0.4 | — | 45.275 | ||
| Molecule 4 | |||||
| LEU63 | 3.8 | — | 45.54 | -10.44EAG| pdb_00004eag | -9.14EAI | pdb_00004eai |
| ARG64 | -4.5 | 12 | 60.113 | ||
| LYS65 | -3.9 | 10.4 | 49.428 | ||
| GLY66 | -0.4 | — | 38.502 | ||
| VAL67 | 4.2 | — | 34.626 | ||
| TYR68 | -1.3 | 10 | 41.507 | ||
| VAL69 | 4.2 | — | 30.89 | ||
| PRO70 | -1.6 | — | 31.656 | ||
| TYR71 | -1.3 | 10 | 31.992 | ||
| THR72 | -0.7 | — | 35.207 | ||
| GLN73 | -3.5 | — | 41.046 | ||
| GLY74 | -0.4 | — | 35.663 | ||
| LYS75 | -3.9 | 10.4 | 39.541 | ||
| TRP76 | -0.9 | — | 30.37 | ||
| GLU77 | -3.5 | 4.3 | 33.203 | ||
| GLY78 | -0.4 | — | 34.057 | ||
| GLU79 | -3.5 | 4.3 | 44.219 | ||
| LEU80 | 3.8 | — | 38.494 | ||
| GLY81 | -0.4 | — | 40.212 | ||
| THR82 | -0.7 | — | 42.033 | ||
| ASP83 | -3.5 | 3.9 | 41.759 | ||
| Molecule 5 | |||||
| PHE47P | 2.8 | — | 39.869 | -9.14EAG| pdb_00004eag | -8.44EAI | pdb_00004eai |
| VAL48P | 4.2 | — | 34.25 | ||
| GLU1 | -3.5 | 4.3 | 41.679 | ||
| MET2 | 1.9 | — | 33.421 | ||
| VAL3 | 4.2 | — | 31.451 | ||
| ASP4 | -3.5 | 3.9 | 35.615 | ||
| ASN5 | -3.5 | — | 28.688 | ||
| LEU6 | 3.8 | — | 30.17 | ||
| ARG7 | -4.5 | 12 | 44.234 | ||
| GLY8 | -0.4 | — | 36.892 | ||
| LYS9 | -3.9 | 10.4 | 43.053 | ||
| SER10 | -0.8 | — | 37.91 | ||
| GLY11 | -0.4 | — | 34.603 | ||
| GLN12 | -3.5 | — | 32.912 | ||
| GLY13 | -0.4 | — | 31.803 | ||
| TYR14 | -1.3 | 10 | 31.279 | ||
| TYR15 | -1.3 | 10 | 32.079 | ||
| VAL16 | 4.2 | — | 33.054 | ||
| GLU17 | -3.5 | 4.3 | 39.069 | ||
| MET18 | 1.9 | — | 34.24 | ||
| THR19 | -0.7 | — | 38.976 | ||
| Molecule 6 | |||||
| PHE47 | 2.8 | — | 46.98 | -9.24EAG| pdb_00004eag | -8.84EAI | pdb_00004eai |
| LEU48 | 3.8 | — | 45.869 | ||
| HIS49 | -3.2 | 6 | 53.639 | ||
| ARG50 | -4.5 | 12 | 51.591 | ||
| TYR51 | -1.3 | 10 | 49.535 | ||
| TYR52 | -1.3 | 10 | 39.934 | ||
| GLN53 | -3.5 | — | 50.401 | ||
| ARG54 | -4.5 | 12 | 44.932 | ||
| GLN55 | -3.5 | — | 57.403 | ||
| LEU56 | 3.8 | — | 54.013 | ||
| SER57 | -0.8 | — | 48.757 | ||
| SER58 | -0.8 | — | 50.077 | ||
| THR59 | -0.7 | — | 46.481 | ||
| TYR60 | -1.3 | 10 | 50.013 | ||
| ARG61 | -4.5 | 12 | 50.64 | ||
| ASP62 | -3.5 | 3.9 | 50.513 | ||
| LEU63 | 3.8 | — | 45.54 | ||
| ARG64 | -4.5 | 12 | 60.113 | ||
| LYS65 | -3.9 | 10.4 | 49.428 | ||
| GLY66 | -0.4 | — | 38.502 | ||
| Molecule 7 | |||||
| TYR68 | -1.3 | 10 | 41.507 | -9.44EAG| pdb_00004eag | -9.14EAI | pdb_00004eai |
| VAL69 | 4.2 | — | 30.89 | ||
| PRO70 | -1.6 | — | 31.656 | ||
| TYR71 | -1.3 | 10 | 31.992 | ||
| THR72 | -0.7 | — | 35.207 | ||
| GLN73 | -3.5 | — | 41.046 | ||
| GLY74 | -0.4 | — | 35.663 | ||
| LYS75 | -3.9 | 10.4 | 39.541 | ||
| TRP76 | -0.9 | — | 30.37 | ||
| GLU77 | -3.5 | 4.3 | 33.203 | ||
| GLY78 | -0.4 | — | 34.057 | ||
| GLU79 | -3.5 | 4.3 | 44.219 | ||
| LEU80 | 3.8 | — | 38.494 | ||
| GLY81 | -0.4 | — | 40.212 | ||
| THR82 | -0.7 | — | 42.033 | ||
| ASP83 | -3.5 | 3.9 | 41.759 | ||
| LEU84 | 3.8 | — | 42.4 | ||
| VAL85 | 4.2 | — | 41.881 | ||
| SER86 | -0.8 | — | 43.472 | ||
| ILE87 | 4.5 | — | 41.825 | ||
| Molecule 8 | |||||
| PRO88 | -1.6 | — | 43.433 | -10.54EAG| pdb_00004eag | -9.84EAI | pdb_00004eai |
| HIS89 | -3.2 | 6 | 47.751 | ||
| GLY90 | -0.4 | — | 46.557 | ||
| PRO91 | -1.6 | — | 49.683 | ||
| ASN92 | -3.5 | — | 57.552 | ||
| VAL93 | 4.2 | — | 51.15 | ||
| THR94 | -0.7 | — | 49.134 | ||
| VAL95 | 4.2 | — | 45.423 | ||
| ARG96 | -4.5 | 12 | 42.488 | ||
| ALA97 | 1.8 | — | 38.478 | ||
| ASN98 | -3.5 | — | 35.288 | ||
| ILE99 | 4.5 | — | 37.365 | ||
| ALA100 | 1.8 | — | 36.58 | ||
| ALA101 | 1.8 | — | 35.89 | ||
| ILE102 | 4.5 | — | 34.453 | ||
| THR103 | -0.7 | — | 37.904 | ||
| GLU104 | -3.5 | 4.3 | 36.466 | ||
| SER105 | -0.8 | — | 34.727 | ||
| ASP106 | -3.5 | 3.9 | 40.665 | ||
| LYS107 | -3.9 | 10.4 | 44.659 | ||
| Molecule 9 | |||||
| GLN73 | -4.5 | 12 | 45.869 | -7.54EAG| pdb_00004eag | -6.94EAI | pdb_00004eai |
| GLY74 | -3.5 | — | 53.639 | ||
| LYS75 | 3.8 | — | 51.591 | ||
| TRP76 | -0.8 | — | 49.535 | ||
| GLU77 | -0.8 | — | 39.934 | ||
| GLY78 | -0.7 | — | 50.401 | ||
| GLU79 | -1.3 | 10 | 43.053 | ||
| LEU80 | -4.5 | 12 | 37.91 | ||
| GLY81 | -3.5 | 3.9 | 34.603 | ||
| THR82 | 3.8 | — | 32.912 | ||
| ASP83 | -4.5 | 12 | 31.803 | ||
| LEU84 | -3.9 | 10.4 | 31.279 | ||
| VAL85 | -0.4 | — | 32.079 | ||
| SER86 | -1.3 | — | 33.054 | ||
| ILE87 | -4.6 | — | 39.069 | ||
| Molecule 10 | |||||
| ILE29 | 3.8 | — | 38.478 | -7.64EAG| pdb_00004eag | -7.14EAI | pdb_00004eai |
| LEU30 | 4.2 | — | 35.288 | ||
| VAL31 | -3.5 | 3.9 | 37.365 | ||
| ASP32 | -0.7 | — | 36.58 | ||
| THR33 | -0.4 | — | 35.89 | ||
| GLY34 | -0.8 | — | 34.453 | ||
| SER35 | -0.8 | — | 37.904 | ||
| Molecule 11 | |||||
| GLY74 | -1.3 | — | 35.663 | -7.14EAG| pdb_00004eag | -6.84EAI | pdb_00004eai |
| LYS75 | -0.7 | 10.5 | 38.541 | ||
| TRP76 | -3.5 | — | 34.37 | ||
| GLU77 | -0.4 | 4.1 | 33.203 | ||
| GLY78 | -3.9 | — | 38.057 | ||
| GLU79 | -0.9 | 4.2 | 40.219 | ||
| LEU80 | -3.5 | — | 38.494 | ||
| GLY81 | -0.4 | — | 41.212 | ||
| THR82 | -3.5 | — | 42.033 | ||
The selection of amino acid residues see table number 4 for molecular interaction or active site analysis primarily depends on several physicochemical and structural parameters, including hydrophobicity, pKa, average isotropic displacement, and molecular docking energy. Each of these parameters provides insight into the stability and binding affinity of ligand–protein complexes. Hydrophobicity represents the tendency of an amino acid to repel water molecules. Positive hydrophobicity values indicate hydrophobic residues (non-polar), such as Leucine, Isoleucine, and Valine, which are typically found in the core or binding pocket of proteins where they enhance hydrophobic interactions with ligands. Negative hydrophobicity values indicate hydrophilic residues (polar or charged), such as Lysine, Aspartic acid, and Glutamic acid, which prefer exposure to aqueous environments and often participate in hydrogen bonding or electrostatic interactions. Therefore, residues with higher (positive) hydrophobicity are generally considered favorable for stable ligand binding due to stronger van der Waals and non-polar interactions. The pKa value of amino acid side chains determines their ionization state at physiological pH. Proper pKa alignment ensures optimal charge complementarity between ligand and protein, enhancing docking efficiency and binding stability. Docking energy reflects the binding affinity between the ligand and protein. Lower (more negative) docking energy indicates stronger and more stable interactions, implying better complementarity between the ligand and amino acid residues in the binding pocket.Thus, residues involved in low docking energy conformations are likely to play key roles in ligand stabilization.
Conclusion
In this study, a series of novel berberine derivatives was designed and computationally evaluated as potential multifunctional inhibitors of AMPK proteins. The present in-silico investigation elucidated the molecular basis of AMPK activation induced by selected bioactive berberine derivatives through multi-target protein interactions relevant to diabetes mellitus. Molecular docking analyses demonstrated that several berberine analogues exhibited superior binding affinities and stable interactions with key metabolic targets, including AMPK, when compared to the parent compound. The hydrogen bonding, hydrophobic, and π–π stacking interactions observed within the active sites confirmed the high affinity and specificity of these derivatives toward their respective receptors. Molecular docking analyses revealed that the derivatives mcles 3 and mcles 4 exhibited stronger binding affinities toward the target protein than the parent molecule, BBR. The results of molecular dynamics simulations further verified that the respective ligand–receptor complexes maintained conformational stability and structural integrity throughout the simulation trajectory, demonstrating consistent interaction patterns within the active site. Comprehensive drug-likeness screening according to the criteria of Lipinski, Ghose, Veber, Egan, and Muegge indicated that several analogues, particularly mcles 3 and mcles 4, fulfilled the majority of physicochemical requirements and possessed favorable bioavailability scores. Overall, the integrated computational findings identify key berberine derivatives with promising therapeutic potential as multitarget modulators for diabetes management. These candidates merit further validation through in vitro enzyme assays and in vivo studies to confirm their efficacy, safety, and mechanism of action. This study provides a rational framework for the design and optimization of next-generation berberine-based antidiabetic therapeutics aimed at AMPK-mediated metabolic regulation.
Acknowledgement
The authors are thankful to Pravara Rural College of Pharmacy, Pravaranagar.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to Reproduce Material from Other Sources
Not Applicable
Author Contributions
Harshali Narayan Anap: Methodology, Writing and Original Draft.
Sunil Eknath Jadhav: In silico ADMET (Absorption, Distribution, Metabolism, Excretion,
Dipali Gorakh Kurkute: In silico ADMET (Absorption, Distribution, Metabolism, Excretion,
Vaijanti Ashok Medge: Calculations of Lipinski’s rule of Five and Druglikeness by Pyrex
Neha Vilas Pagare: Docking images of 2D and 3D by Discovery Studio 2021 software
Soniya Vaibhav Katore: The active amino residues, bond length, bond category, bond type, ligand energies, and docking scores.
Rohit Jaysing Bhor: Methodology, Writing and Original Draft.
Sujata Eknath Nirmal: Pyrex Software’s and SWISS ADMET Software
Jyoti Ramkisan Bhagat: The active amino residues, bond length, bond category, bond type, ligand energies, and docking scores.
References
- Daousi, C. (2006). Prevalence of obesity in type 2 diabetes in secondary care: Association with cardiovascular risk factors. Postgraduate Medical Journal, 82(966), 280–284. https://doi.org/10.1136/pmj.2005.039032
CrossRef - Ma, H., He, K., Zhu, J., Li, X., & Ye, X. (2019). The anti-hyperglycemia effects of Rhizoma Coptidis alkaloids: A systematic review of modern pharmacological studies of the traditional herbal medicine. Fitoterapia, 134, 210–220. https://doi.org/10.1016/j.fitote.2019.03.003
CrossRef - Kong, Y., Li, L., Zhao, L. G., Yu, P., & Li, D. D. (2021). A patent review of berberine and its derivatives with various pharmacological activities (2016–2020). Expert Opinion on Therapeutic Patents, 1–13. https://doi.org/10.1080/13543776.2021.1974001
CrossRef - Ye, Y., Liu, X., Wu, N., et al. (2021). Efficacy and safety of berberine alone for several metabolic disorders: A systematic review and meta-analysis of randomized clinical trials. Frontiers in Pharmacology, 12. https://doi.org/10.3389/fphar.2021.653887
CrossRef - Liang, Y., Xu, X., Yin, M., et al. (2019). Effects of berberine on blood glucose in patients with type 2 diabetes mellitus: A systematic literature review and a meta-analysis. Endocrine Journal, 66(1), 51–63. https://doi.org/10.1507/endocrj.EJ18-0109
CrossRef - Ju, J., Li, J., Lin, Q., & Xu, H. (2018). Efficacy and safety of berberine for dyslipidaemias: A systematic review and meta-analysis of randomized clinical trials. Phytomedicine, 50, 25–34. https://doi.org/10.1016/j.phymed.2018.09.212
CrossRef - Sanjari, M., Shamsinejad, B., Khazaeli, P., Safi, Z., Mirrashidi, F., & Naghibzadeh-Tahami, A. (2020). Safety and efficacy of Berberis integerrima root extract in patients with type 2 diabetes: A parallel intervention-based triple blind clinical trial. Journal of Diabetes & Metabolic Disorders, 19(1), 71–80. https://doi.org/10.1007/s40200-019-00478-z
CrossRef - Zhang, Y., Li, X., Zou, D., et al. (2008). Treatment of type 2 diabetes and dyslipidemia with the natural plant alkaloid berberine. The Journal of Clinical Endocrinology & Metabolism, 93(7), 2559–2565. https://doi.org/10.1210/jc.2007-2404
CrossRef - Lan, J., Zhao, Y., Dong, F., et al. (2015). Meta-analysis of the effect and safety of berberine in the treatment of type 2 diabetes mellitus, hyperlipemia and hypertension. Journal of Ethnopharmacology, 161, 69–81. https://doi.org/10.1016/j.jep.2014.09.049
CrossRef - Liu, Y., et al. (2023). Berberine stimulates lysosomal AMPK independent of PEN2 and UHRF1-mediated dephosphorylation. Frontiers in Pharmacology, 14, 114861. https://doi.org/10.3389/fphar.2023.1148611
CrossRef - Li, Y., et al. (2023). A mechanistic review on how berberine use combats metabolic disorders. Pharmaceutics, 17(1), 7. https://doi.org/10.3390/pharmaceutics17010007
CrossRef - Roy, A., et al. (2025). Unveiling berberine analogues as potential inhibitors: Virtual screening, molecular docking, and ADMET profiling. Scientific Reports. https://doi.org/10.1038/s41598-025-98835-x
CrossRef - De Oliveira, V. M., et al. (2024). Insights from structure-based virtual screening of aryl-substituted berberine-benzimidazole derivatives. Journal of Molecular Recognition. (In Press). https://doi.org/10.1002/jmr.####
- Mohanty, S., et al. (2020). Berberine improves glucose and lipid metabolism in HepG2 cells through AMPKα1 activation. Frontiers in Pharmacology, 11, 647. https://doi.org/10.3389/fphar.2020.00647
CrossRef - Zhang, W., et al. (2024). Molecular mechanisms, targets and clinical potential of berberine in metabolism and chronic disease. Frontiers in Pharmacology, 15, 1368950. https://doi.org/10.3389/fphar.2024.1368950
CrossRef - Zhu, X., et al. (2023). Natural products targeting AMPK signalling pathway therapy: Progress and perspectives. Frontiers in Pharmacology, 16, 1534634. https://doi.org/10.3389/fphar.2025.1534634
CrossRef - Zhang, X., et al. (2022). Network pharmacology and molecular docking analysis on targets of berberine against metabolic syndrome. Journal of Integrative Medicine, 20(1), 1–13. https://doi.org/10.1016/j.joim.2021.11.001
CrossRef - Mohanty, P., et al. (2023). Boldine and berberine in silico molecular docking studies of their metabolites for antidiabetic potential. Pharmaceutics, 15(10), 1195. https://doi.org/10.3390/pharmaceutics15101195
CrossRef - Ooi, S. K., et al. (2024). Berberine modulates cardiovascular and metabolic diseases as a multitarget alkaloid: An in-silico screening approach. Pharmacology Research, 180, 106343. https://doi.org/10.1016/j.phrs.2024.106343
- Wang, J., et al. (2023). Addressing the preventive and therapeutic perspective of berberine against diabetes: A review. Heliyon, 9(4), e08441. https://doi.org/10.1016/j.heliyon.2023.e08441
CrossRef - Davis, L., & Patel, B. (2025). In silico strategies for drug discovery: Optimizing natural compounds derived from food. Natural Compound Drug Discovery, 12(2), 45–68. https://doi.org/10.1007/s44371-025-00201-3
CrossRef - Zhao, Y., et al. (2023). In silico exploration of berberine as a potential ADME-toxicity agent using network pharmacology and molecular docking. International Journal of Applied Pharmacology, 15(8), 49922.
Abbreviations List
GLU: Glutamic acid; THR: Threonine; LEU: Leucine; GLN:Glutamine; VAL: Valine; HIS: Histidine; ASP: Aspartic acid GLY: Glycine; LYS: Lysine; ILE: Isoleucine; ASN: Asparagine; ALA: Alanine; PRO: Proline; SER: Serine; TYR; Tyrosine; GI absorption Gastrointestinal absorption; BBB: Blood–brain barrier; Pgp: P-glycoprotein; CYP: Cytochrome P450 enzyme; CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4: Specific isoforms of Cytochrome P450 enzymes involved in drug metabolism; log Kp: Skin permeation coefficient (cm/s); Lipinski violations: Number of violations of Lipinski’s Rule of Five (drug-likeness rule); ADMET: Absorption, Distribution, Metabolism, Excretion, and Toxicity; TPSA: Topological polar surface area; Log P: Octanol–water partition coefficient; SMILES: Simplified Molecular Input Line Entry System; 3IHZ: PDB ID of Plasmodium vivax protease used as the target protein
Accepted on: 26-11-2025
Second Review by: Dr. Akshaya Arva
Final Approval by: Dr. Wagih Ghannam








