
Computationally Guided Design and Synthesis of Quinazoline Thiazolidinone Scaffolds Targeting Fructose 1, 6-Bisphosphatase
 , Sanjay Sawant1, Komal Lahoti1, Shankar Chavan1, Aditi Pawar1, Rushikesh Yande1, Harshal Waghmare1and Minal Ghante2
, Sanjay Sawant1, Komal Lahoti1, Shankar Chavan1, Aditi Pawar1, Rushikesh Yande1, Harshal Waghmare1and Minal Ghante2 1Department of Pharmaceutical Chemistry, STES, Smt. Kashibai Navale College of Pharmacy, Savitribai Phule Pune University, Maharashtra, India.
2Department of Pharmaceutical Quality Assurance, STES, Smt. Kashibai Navale College of Pharmacy, Savitribai Phule Pune University, Maharashtra, India.
Corresponding Author E-mail: pr.vasundhara@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3493
Download this article as:
![]()
Human fructose-1,6-bisphosphatase (FBPase), a key regulatory enzyme of hepatic gluconeogenesis, represents an important therapeutic target for the management of type 2 diabetes mellitus. However, classical phosphate-based FBPase inhibitors are often associated with poor pharmacokinetic properties. In the present study, a series of novel quinazolinone–thiazolidin-4-one hybrid derivatives (PT-01 to PT-10) were rationally designed as non-phosphate FBPase inhibitors, synthesized, and structurally confirmed by FT-IR, ¹H NMR, mass spectrometry and elemental analysis. Molecular docking studies were carried out using Auto Dock software against the crystal structure of human FBPase (PDB: 6LS5) to evaluate binding within the allosteric adenine-binding pocket. The synthesized compounds exhibited docking scores ranging from –7.3 to –9.7 kcal/mol, demonstrating stronger predicted binding affinity than the reference inhibitor CS-917 (–6.9 kcal/mol). Key interactions involved residues Val17, Ala24, Leu30, Met177, Arg140, Thr31, and Tyr113. Substituent variation significantly influenced binding, with electron-donating and electron-withdrawing groups enhancing hydrogen bonding and hydrophobic stabilization. Among the series, PT-04 (m-nitrophenyl substituted) showed the highest docking score (–9.7 kcal/mol) and stable multipoint interactions. In a 28-day in vivo evaluation using a diabetic rodent model, the selected compound PT-03 produced a reduction in blood glucose levels from 507 mg/dL to 302 mg/dL, indicating a measurable anti hyperglycaemic effect. The In silico ADMET and drug-likeness analyses suggested acceptable pharmacokinetic properties and compliance with Lipinski’s criteria. Collectively, these findings identify the quinazolinone–thiazolidinone scaffold as a viable non-phosphate framework for further optimization and detailed pharmacological investigation as FBPase-targeted antidiabetic agents.
KEYWORDS:Fructose-1; 6-bisphosphatase; Gluconeogenesis; Quinazoline; Thiazolidinone; Type 2 Diabetes Mellitus
Introduction
Type 2 diabetes mellitus (T2DM) mainly occur due to insufficient secretion of insulin, reduced glucose uptake, elevated glucose production and/or insulin resistance. This ultimately results in fasting and postprandial hyperglycaemia.1–3 According to the International Diabetes Federation (IDF) Diabetes Atlas (2023), approximately 537 million adults worldwide are affected by diabetes, with projections rising to 643 million by 2030 and 783 million by 2045; nearly 90–95% of these cases are T2DM.4,5 Although current therapies improve insulin secretion, enhance insulin sensitivity, delay glucose absorption, or promote renal glucose excretion, only metformin significantly suppresses hepatic gluconeogenesis.6–8 Earlier studies reveal that, in T2DM gluconeogenesis mediated endogenous glucose overproduction plays crucial role and till date only metformin acts via this mechanism to effectively control the hyperglycaemia.9 Since excessive gluconeogenesis plays a central role in endogenous glucose overproduction in T2DM, direct targeting of this pathway represents a promising but underexploited therapeutic strategy.10,11
Fructose-1, 6-bisphosphatase (FBPase) catalyses the rate determining step in middle phase of gluconeogenesis process in which fructose-1, 6-bisphosphate is transformed into fructose-6-phosphate.12–15 FBPase is a homo-tetramer, and it exists in two forms viz. active (R) and inactive (T) conformational states.15,16,17 At present, many pharmaceutical industries and academic researchers are involved in developing the novel and effective allosteric inhibitors against FBPase.18–20 amongst them only CS-917 and MB07803 were progressed into phase II clinical trial.21–25 Many of the compounds failed during clinical trials primarily due to unfavourable interactions of phosphonate group at an active site of FBPase. This structural feature also resembles with AMP causing interference in other metabolic pathways and leading to some side effects.26–28 In 2008, the clinical trial of the CS- 917, phosphonic diamide prodrug was halted due to lactic acidosis.20,29 Therefore, it is thought-provoking to develop non-AMP mimetic safe and effective FBPase inhibitors. In the quest of searching non phosphorous containing novel FBPase inhibitors, nitrogenous heterocycle substituted with thiazolidinone ring was chosen for study and thoroughly structural features required for activity are examined. Prompted by aforementioned facts, in this research work, novel FBPase inhibitors were designed by the integration of the quinazolin-4-one and thiazolidinone rings and synthesized with a goal to improve the binding affinity and minimizing unfavourable interactions at an active site. 30,31
Nitrogen containing heterocycles such as quinazolin-4-one and thiazolidin-4-one exhibit diverse pharmacological activities, including antidiabetic and enzyme inhibitory effects, and possess favorable structural features for enzyme binding through hydrogen bonding and π–π interactions.32,33 Despite their potential, quinazolinone thiazolidinone hybrids have been scarcely explored as non-phosphorus FBPase inhibitors. Therefore, the present study addresses this gap by designing and synthesizing novel quinazolinone thiazolidinone hybrid derivatives as non-AMP mimetic FBPase inhibitors. The objectives were to synthesize and characterize the compounds, evaluate their biological activity, analyze structure activity relationships, and perform molecular docking studies to enhance binding affinity while minimizing unfavorable active-site interactions, thereby establishing a rational and potentially safer design strategy.
Materials and Methods
All the reagents and chemicals used for the study are procured from Sigma Aldrich, Loba Chemie, Pvt. Ltd. Mumbai, India; and HiMedia Laboratory Pvt. Ltd., Delhi, India. These are used without further purification process. Thin layer chromatography was used to monitor the progress of reaction. The progress of all reactions was monitored by thin layer chromatography (TLC) using Merck pre-coated silica gel G plates with a layer thickness 175-225 µm and different eluent systems used were Ethyl acetate: n-Hexane: Ethanol (6:3:1 v/v/v). The spots of compounds on TLC plates were visualized by UV light irradiation at long range wavelengths between 315-400 nm and for shorter wavelength between 100-280 nm. All the synthesized compounds were studied by melting point using open capillary tube method. Bruker Avance 400 model was used to record 1H-NMR spectra at 400 MHz in DMSO using tetramethyl silane (internal standard). The splitting pattern abbreviations used is as follows: s for singlet, d for doublet and m for multiplet. The IR spectra were recorded on a Bruker Alpha II model spectrophotometer using KBr-pellet method. The Mass spectra were recorded using WATERS 3100 model of MS ES+ and ES- (300-800 m/z) mass spectrometer.
General procedure of the synthesized compounds
Synthesis of 6-Fluoro-2-phenyl-4H-benzo[d] [1, 3] oxazin-4-one (1)
5mM of 2-Amino-5-fluorobenzoic acid was dissolved in 30 ml of pyridine in a flask. Then 2.5 ml of benzoyl chloride was added drop wise with continuous stirring for 5 minutes. Reaction mixture was set aside for 25 minutes at room temperature (RT) with occasional shaking and then poured into cold water to form a precipitate. The precipitate thus formed was filtered and washed with 5% NaHCO3 and dried at 100oC followed by recrystallization from ethanol. Yield: 55.4%, Rf = 0.8.
Synthesis of 6-Fluoro-3-amino-2-phenyl-3, 4-dihydroquinazolin-4-one (2)
Compound (1) (5mM) was dissolved in 2 ml of 80% hydrazine hydrate. To this mixture 7 ml of pyridine was added and then the mixture was refluxed for 6 hours at 80oC. After completion of the reaction, the mixture was cooled at RT and crushed ice was added to obtain a solid mass. After washing with 5% NaCl and NaHCO3, the product was dried and recrystallized from ethanol. Yield: 63.1%, Rf = 0.8.
General procedure for synthesis of 6-Fluoro-3- {(2-phenyl, substituted)-4(oxothiazolidin-3-yl)}-2-phenylquinazolin-4(3H)-one (PT-01 to PT-10 Compounds)
Aromatic/aliphatic aldehyde (0.05 mM), 25 ml of dimethylformamide (DMF) and 2-3 drops of glacial acetic acid was added in compound (2). To this mixture 1.84 ml (4mM) of thioglycolic acid (TGA) was added with constant stirring at 00C. After 5 minutes, 2 mM of N, N`-dicyclohexylcarbodiimide (DCC) was added to reaction mixture. The resulting mixture was stirred for 5 hours, and then dicyclohexylurea was filtered off. The final product was obtained upon addition of 25 ml of cold water to the filtrate. The corresponding crystalline product was recrystallized with ethanol.
Spectral characteristics of the synthesized compounds
Synthesis of 6-fluoro-3-(4-oxo-2-phenylthiazolidin-3-yl)-2-phenylquinazolin-4(3H)-one (PT-01). Yield: 61%; M.P. 164-168oC, Rf =0.7. IR (KBr cm-1); 1182 (N–N), 1367 (C–N), 1105 (C–F), 1629 (C=N), 779 (C–S–C), 1688 (amide C=O), 3032 (Ar–C–H), 1516 (aromatic C=C). 1H-NMR (DMSO, δ, ppm): 7.9–8.5 (m, 10H, aromatic protons), 3.60–3.72 (s, 2H, CH₂ of thiazolidinone), 3.25–3.35 (s, 1H, CH of thiazolidinone). ESI MS (m/z): 419.6 (M+2H)+.
Synthesis of 6-fluoro-3-(2-(4-hydroxyphenyl)-4-oxothiazolidin-3-yl)-2-phenylquinazolin-4(3H)-one (PT-02). Yield: 56%; M.P. 120-125oC, Rf =0.8. IR (KBr cm-1); 1089 (N–N), 1310 (C–N), 1110 (C–F), 1689 (C=N), 776 (C–S–C), 1627 (amide C=O), 3520 (O–H), 3030 (Ar–C–H), 1513 (Ar C=C). 1H-NMR (DMSO, ẟ, ppm): 7.6–8.5 (m, 9H, aromatic), 5.30 (s, 1H, phenolic OH), 3.60–3.75 (s, 2H, CH₂), 3.20–3.30 (s, 1H, CH). ESI MS (m/z): 433.4 (M)+.
Synthesis of 3-(2-(4-(dimethyl amino) phenyl)-4-oxothiazolidin-3-yl)-6-fluoro-2-phenylquinazolin-4(3H)-one (PT-03). Yield: 53%; M.P. 123-126oC, Rf =0.5. IR (KBr cm-1); 1159 (N–N), 1314 (C–N), 1112 (C–F), 1667 (C=N), 771 (C–S–C), 1627 (amide C=O), 3030 (Ar–C–H), 1574 (Ar C=C). 1H-NMR (DMSO, ẟ, ppm): 7.8–8.5 (m, 9H, aromatic), 3.00 (s, 6H, N(CH₃)₂), 3.62–3.75 (s, 2H, CH₂), 3.25 (s, 1H, CH). ESI MS (m/z): 461.4 (M+H)+.
Synthesis of 6-fluoro-3-(2-(2-nitrophenyl)-4-oxothiazolidin-3-yl)-2-phenylquinazolin-4(3H)-one (PT-04). Yield: 67 %; M.P. 107⁰C-111⁰C, Rf = 0.4. IR (KBr, cm-1); 1230 (N–N), 1309 (C–N), 1108 (C–F), 1689 (C=N), 737 (C–S–C), 1524 & 1348 (NO₂), 1627 (amide C=O), 3030 (Ar–C–H). 1H-NMR (DMSO, δ, ppm): 7.6–8.9 (m, 9H, aromatic including nitrophenyl), 3.60–3.72 (s, 2H, CH₂), 3.20 (s, 1H, CH). ESI MS (m/z): 461.0 (M)+.
Synthesis of 6-fluoro-3-(2-isopropyl-4-oxothiazolidin-3-yl)-2-phenylquinazolin-4(3H)-one (PT-05). Yield: 71%; M.P. 118⁰C-120⁰C, Rf = 0.8. IR (KBr, cm-1); 1156 (N–N), 1245 (C–N), 1102 (C–F), 1671 (C=N), 734 (C–S–C), 1628 (amide C=O), 2960–2870 (alkyl C–H), 3030 (Ar–C–H). 1H-NMR (DMSO, δ, ppm): 7.4–8.2 (m, 8H, aromatic), 1.10–1.25 (d, 6H, isopropyl CH₃), 2.85–2.95 (m, 1H, isopropyl CH), 3.60–3.70 (s, 2H, CH₂), 3.20 (s, 1H, CH). ESI MS (m/z): 382.2 (M)+.
Synthesis of 6-fluoro-3-(2-(furan-2-yl)-4-oxothiazolidin-3-yl)-2-phenylquinazolin-4(3H)-one (PT-06). Yield: 51.2%; M.P. 131⁰C-133⁰C, Rf = 0.7. IR (KBr, cm-1); 1244 (N–N), 1311 (C–N), 1108 (C–F, aryl), 1676 (C=N), 755 (C–S–C), 1627 (amide C=O), 1244 (C–O–C, furan), 3030 (Ar–C–H), 1576 (aromatic C=C). 1H-NMR (DMSO, δ, ppm): 7.40–8.20 (m, 8H, aromatic protons), 6.35–7.25 (m, 3H, furan protons), 3.60–3.72 (s, 2H, CH₂ of thiazolidinone), 3.20–3.30 (s, 1H, CH of thiazolidinone) . ESI MS (m/z): 407.8 (M)+.
Synthesis of 6-fluoro-3-(2-(4-methoxyphenyl)-4-oxothiazolidin-3-yl)-2-phenylquinazolin-4(3H)-one (PT-07). Yield: 69.7%; M.P. 159⁰C-163⁰C, Rf = 0.9. IR (KBr, cm-1); 1174 (N-N), 1311 (C-N), 1605 (C=N), 690 (C-Cl), 1029 (C-O), 2929 (Ar, C-H), 1672 (Amide, CONH), 1572 (Benzene). 1H-NMR (DMSO, δ, ppm): 7.50–8.30 (m, 9H, aromatic protons), 3.75 (s, 3H, OCH₃), 3.60–3.70 (s, 2H, CH₂ of thiazolidinone), 3.25–3.35 (s, 1H, CH of thiazolidinone), ESI-MS (m/z): 449.3 (M+2H)+.
Synthesis of 3-(2-(2-chlorophenyl)-4-oxothiazolidin-3-yl)-6-fluoro-2-phenylquinazolin-4(3H)-one (PT-08). Yield: 70.4%; M.P. 216⁰C-218⁰C, Rf = 0.6. IR (KBr, cm-1); 1032 (N–N), 1315 (C–N), 1107 (C–F, aryl), 1627 (C=N), 761 (C–S–C), 1687 (amide C=O), 3030 (Ar–C–H), 1517 (aromatic C=C), 760 (C–Cl, aryl). 1H-NMR (DMSO, δ, ppm): 7.20–8.30 (m, 9H, aromatic protons), 3.60–3.72 (s, 2H, CH₂ of thiazolidinone), 3.30–3.40 (s, 1H, CH of thiazolidinone). ESI MS (m/z): 452.6 (M+H)+.
Synthesis of 6-fluoro-3-(2-(2-hydroxyphenyl)-4-oxothiazolidin-3-yl)-2-phenylquinazolin-4(3H)-one (PT-09). Yield: 71.7%; M.P. 180⁰C-182⁰C, Rf = 0.8. IR (KBr, cm-1); 1156 (N–N), 1309 (C–N), 1109 (C–F, aryl), 1626 (C=N), 751 (C–S–C), 3327 (O–H, phenolic), 1661 (amide C=O), 3030 (Ar–C–H), 1513 (aromatic C=C). 1H-NMR (DMSO, δ, ppm): 7.40–8.30 (m, 9H, aromatic protons), 5.40 (s, 1H, phenolic OH), 3.60–3.72 (s, 2H, CH₂ of thiazolidinone), 3.25–3.35 (s, 1H, CH of thiazolidinone). ESI MS (m/z): 435.9 (M+2H)+.
Synthesis of 3-(2-(4-chlorophenyl)-4-oxothiazolidin-3-yl)-6-fluoro-2-phenylquinazolin-4(3H)-one (PT-10). Yield: 58.8 %; M.P. 115⁰C-119⁰C, Rf = 0.8. IR (KBr, cm-1); 1245 (N–N), 1312 (C–N), 1106 (C–F, aryl), 1627 (C=N), 735 (C–S–C), 1669 (amide C=O), 3030 (Ar–C–H), 1575 (aromatic C=C), 755 (C–Cl, aryl). 1H-NMR (DMSO, δ, ppm): 7.50–8.40 (m, 9H, aromatic protons), 3.60–3.75 (s, 2H, CH₂ of thiazolidinone), 3.30–3.40 (s, 1H, CH of thiazolidinone). ESI MS (m/z): 452.8 (M+H)+.
Computational Studies
The target protein (FBPase) fructose 1,6-bisphosphatase (PDB ID: 6LS5) was selected for this research work based on resolution of 2.03A0. It is isolated from human liver and characterized by X-ray diffraction method. 15 The crystal structure of target enzymatic protein, Fructose 1,6-bisphosphatase was downloaded from protein data bank (PDB ID: 6LS5) available at Research Collaboratory Structural Bioinformatics (RCSB) website. (www.rcsb.org) For an active site, the grid dimensions were used as (x = -18.436304, y = 60.330783, z = -16.143913) and size of the grid box was set to (40 Å × 40 Å × 40 Å). Exhaustiveness was used as 8 during this process. Library of various ligand molecules were designed based on the rationale of structure-based drug design. The 2D structures of designed ligand molecules were drawn using ChemDraw Ultra 8.0 tool and were converted to 3D structures through energy minimization process. The protein structure was prepared using Biovia Discovery Studio and AutoDock tools by removal of water molecules, repairing the missing atoms and Kollman and Gasteiger charges were assigned. The active site of protein structure was validated followed by minimization of 0.3Å through root-mean-square deviation was done. The energy minimization process was done with the AutoDockTools-1.5.6 using force field (MMFF). Molecular docking studies were accomplished and results were evaluated by respective docking scores with RMSD values of the best pose.34,35
In silico ADME prediction
The web tools viz. Swiss ADME webserver and ADMET lab 3.0 were used to analyse the compounds for drug likeness profile. The various drug likeness rules taken into consideration for the study including Lipinski’s, Veber, Ghose, Egan’s and Muegge’s rule.36
Biological evaluation
Induction of experimental diabetes
The dose of 60 mg/kg streptozotocin was given to overnight fasted animals intra-peritoneally. Blood glucose levels were monitored after 48 h of administration. Blood glucose level more than 250 mg/dL after 48 h of STZ injection was noted to be diabetic in animals during the study.37
Experimental protocol to study In vivo glucose lowering effect
For this study, 3 group of 6 animals each were prepared viz. diabetic control (Group 1) treated with 0.5% (w/v) aqueous CMC solution (5 mL/kg). Standard (Group 2) with treatment of 10 mg/kg in metformin 0.5% (w/v) aqueous CMC and Test (Group 3) treated with 100 mg/kg of the test compound. The blood glucose levels were monitored at 7 days intervals starting from Day 0. Blood sample was collected from retroorbital plexus.
During the experimental period, animals were observed daily for signs of toxicity or behavioural changes, including lethargy, tremors, piloerection, altered locomotion, or abnormal posture. Body weight was recorded at baseline and weekly thereafter. Food and water intake were measured daily (or every 2–3 days) per cage. Mortality, if any, was recorded throughout the study duration. These parameters were monitored to evaluate the systemic safety and tolerability of the test compounds.
Fructose-1,6-bisphosphatase (FBPase) was selected as the molecular target because it catalyzes a rate-limiting step in hepatic gluconeogenesis and has been extensively validated as a therapeutic target for reducing excessive hepatic glucose production. Although hyperglycaemia in streptozotocin induced diabetic mice is primarily driven by insulin deficiency due to pancreatic β-cell damage, increased hepatic gluconeogenesis is also a significant contributing factor in this model. Therefore, FBPase inhibition may still partially attenuate hyperglycaemia by reducing endogenous glucose output. However, we acknowledge that the STZ model more closely mimics type 1 diabetes, whereas FBPase inhibition is particularly relevant in type 2 diabetes characterized by insulin resistance and dysregulated gluconeogenesis. Future studies using insulin-resistant or diet-induced diabetic models would provide a more physiologically relevant evaluation of FBPase-targeted therapy.
Sample Size Limitation
We acknowledge that the use of only two animals in one experimental group represents a limitation of the present study. The in-vivo experiment was conducted as a preliminary screening to assess proof-of-concept antihyperglycemic activity of selected lead compounds. Due to logistical and ethical constraints, the sample size was limited; therefore, the results should be interpreted as exploratory rather than definitive. Larger, statistically powered studies with appropriate group sizes will be necessary to confirm the observed glucose-lowering effects and to establish reproducibility and significance.
Data analysis using statistical methods
Using graph pad prism 6.0 software statistically data was analysed and results were recorded as mean ± SD. One-way ANOVA with student’s t-test method was used in data analysis and p values < 0.05 indicate statistical significance. The values were significant at p < .05 and p < .01 level.
Study Design
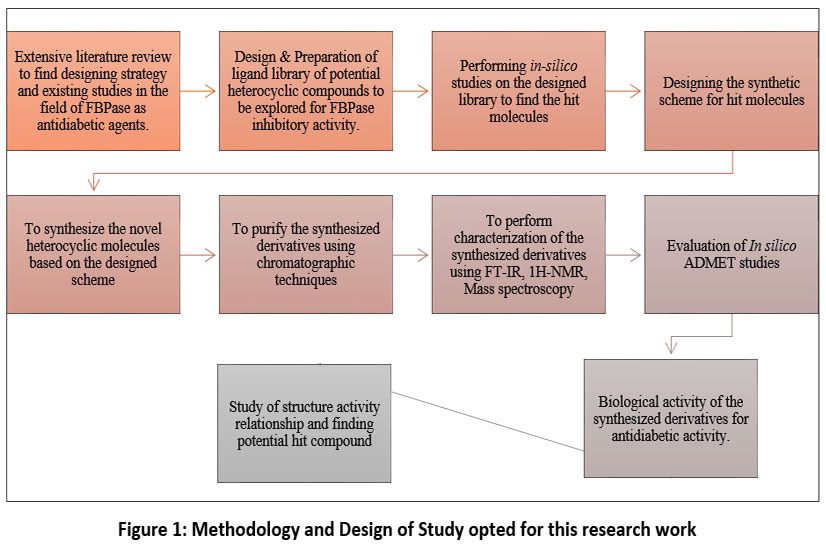 |
Figure 1: Methodology and Design of Study opted for this research work
|
The Figure 1 represents the design of study carried out to perform this research work.
Results
Rationale for design of compounds
Fructose-1, 6-bisphosphatase (FBPase) is a key regulatory enzyme in hepatic gluconeogenesis, and its inhibition has emerged as a validated strategy for controlling fasting hyperglycemia in type 2 diabetes. The rational design of small-molecule FBPase inhibitors requires scaffolds capable of engaging a polar, metal-dependent catalytic site while maintaining favourable pharmacokinetic properties. The quinazolinone–thiazolidinone hybrid scaffold fulfils these requirements through complementary structural and electronic features. The present design strategy is based on the rational hybridization of quinazolinone and thiazolidinone pharmacophores, both of which are well recognized for their enzyme-binding capability and antidiabetic relevance. The quinazolin-4(3H)-one core was selected as the primary anchoring unit due to its planar heteroaromatic framework and the presence of hydrogen-bond-accepting nitrogen atoms and a lactam carbonyl group, which favour strong interactions within the FBPase binding pocket. Substitution at the R position enables modulation of electronic and lipophilic properties to optimize target affinity.
The 4-oxothiazolidinone moiety was incorporated to complement the quinazolinone core, providing additional polar functionality and conformational adaptability. The carbonyl and sulphur-containing features of the thiazolidinone ring are expected to enhance interactions with catalytically important residues of FBPase, which operates through a metal-dependent mechanism. Phenyl substitution attached to this moiety allows further tuning of hydrophobic and π–π interactions. The hybrid scaffold was designed to promote multi-point binding within the enzyme site, thereby enhancing inhibitory potential while maintaining drug-like physicochemical characteristics. This strategy provides a flexible platform for systematic SAR exploration and supports the development of novel FBPase-targeted antidiabetic agents.
At the allosteric site of fructose-1, 6-bisphosphatase (FBPase), the adenine-binding hydrophobic pocket is primarily defined by key amino acid residues including Val17, Ala24, Leu30, and Met177, which play a crucial role in ligand stabilization. In addition, strong conventional hydrogen-bonding interactions have been reported between the phosphate moiety of endogenous ligands and active-site residues such as Glu29, Gly28, Thr27, Lys112, and Tyr113, highlighting the importance of polar interactions in enzyme inhibition. However, despite the significant contribution of phosphate groups to binding affinity, phosphate- and phosphonate-based inhibitors often exhibit poor pharmacokinetic properties, including low membrane permeability and unfavourable oral bioavailability, thereby limiting their therapeutic development.
In view of these limitations, the present study was directed toward the design of non-phosphate FBPase inhibitors capable of retaining key binding interactions while improving drug-like characteristics. Accordingly, quinazolinone-linked thiazolidin-4-one derivatives were rationally designed and synthesized as alternative scaffolds for FBPase inhibition. Among the designed series, Scaffold A was selected as the core template for systematic structural modification and subsequent structure–activity relationship (SAR) optimization.
At an allosteric site of FBPase adenine binding hydrophobic pocket has been aligned with key amino acids such as Val17, Ala24, Leu30, and Met177. Strong conventional hydrogen bonding interactions were observed between phosphate group and active site residues like, Glu29, Gly28, Lys112, Thr27 and Tyr113. Phosphate group primarily contributes for the binding affinity at an active site however the compounds with phosphonate suffer from poor pharmacokinetic profile for further development.38–41 Hence, this study intended to explore enzyme inhibitors with other than phosphate functional groups. Hence, quinazoline clubbed thiazolidine-4-one derivatives were designed and synthesized as FBPase inhibitors.
Table 1. Docking studies and binding mode analysis at the active site of Fructose 1, 6-bisphophatase
| Compound | Substituent | Docking Score | H Bonding Interactions | Other Interactions |
| PT01 | Phenyl | -7.3 | THR31 | MET 177, LEU 30, ALA 24, GLU 20 |
| PT02 | p-Hydroxyphenyl | -7.6 | ARG 140 | LEU 30, ALA 24, ARG 140, GLU 20, LYS 23 |
| PT03 | p-dimethyl aminophenyl | -9.2 | ARG 140 | GLU 20, MET 177, LEU 30, GLY 28, CYS 179, ALA24
ARG25 |
| PT04 | m-Nitrophenyl | -9.7 | ARG140, THR31, TYR113 | ARG140, ALA24, LEU30, GLY26, GLY21, ARG25 |
| PT05 | -Isopropyl | -7.5 | THR 31: 2.33, GLY 28: 2.58, ARG 140: (2.25, 1.99) | ALA 24,MET 117, CYS 179, LEU 30 |
| PT06 | Furan-2-yl | -7.9 | THR 31: 2.52 | MET 177, LEU 30, ALA 24, ARG 140, GLU 20, GLY 21 |
| PT07 | p-Methoxyphenyl | -8.9 | ARG 140: (2.25, 1.99) | MET 177, ALA 24,LEU 30, VAL 17 |
| PT08 | o-Chlorophenyl | -7.7 | ARG 140: 2.47 | MET 177, LEU 30, ALA 24, CYS 179, GLU 20 |
| PT09 | o-Hydroxyphenyl | -8.1 | ARG 140: (2.45, 2.19) | MET 177, ALA 24, LEU 30, VAL 17 |
| PT10 | p-Chlorophenyl | -7.3 | THR 31, GLY 28 | THR 31, GLY 28, LEU 30, ARG 140, ALA 24, VAL 160, ASP 178 |
| CS 917 (Reference) | – | -6.9 | ALA 24: 2.32, LYS 112: 2.05 | CYS 179, GLY 26, LEU 30, MET 177, ARG 140 |
Molecular docking studies were performed to elucidate the binding behaviour of the synthesized quinazoline–thiazolidinone derivatives (PT01–PT10) within the fructose-1, 6-bisphosphatase (FBPase) binding pocket and to rationalize the influence of structural variations on binding affinity. (Table 1) The docking scores of the compounds ranged from –7.3 to –9.7 kcal/mol, with several derivatives exhibiting significantly improved binding compared to the reference inhibitor CS-917 (–6.9 kcal/mol).
Substituent-dependent variations significantly influenced binding affinity across the series. PT01 with unsubstituted phenyl showed modest interaction (–7.3 kcal/mol), mainly through hydrophobic contacts with Met177, Leu30, and Ala24, while polar substitutions such as PT02 and PT09 moderately improved affinity via hydrogen bonding with Arg140. Electron-donating groups markedly enhanced activity, as observed with PT03 (p-dimethylamino, –9.2 kcal/mol) and PT07 (p-methoxy, –8.9 kcal/mol), reflecting favorable electronic effects and improved electrostatic stabilization within the binding pocket. Notably, PT04 bearing a m-nitro substituent exhibited the strongest binding (–9.7 kcal/mol), forming multiple hydrogen bonds with Arg140, Thr31, and Tyr113 along with extensive hydrophobic interactions, thereby producing a highly stabilized complex.
Molecular Docking and Structure-Binding Relationship
Substituent-dependent trends were clearly observed. PT-01 (phenyl) and PT-10 (p-chlorophenyl) showed moderate binding (–7.3 kcal/mol), mainly through hydrophobic interactions with Ala24, Leu30, and Met177. Introduction of hydroxyl groups improved affinity, as seen with PT-02 (–7.6 kcal/mol) and PT-09 (–8.1 kcal/mol), which formed hydrogen bonds with Arg140, highlighting the importance of polar interactions. Electron-donating substituents further enhanced activity; PT-03 (p-dimethyl amino, –9.2 kcal/mol) and PT-07 (p-methoxy, –8.9 kcal/mol) demonstrated strong binding through hydrogen bonding with Arg140 and multiple hydrophobic contacts within the pocket. In contrast, the aliphatic PT-05 (isopropyl, –7.5 kcal/mol) showed reduced affinity, underscoring the role of aromaticity in stabilizing enzyme interactions.
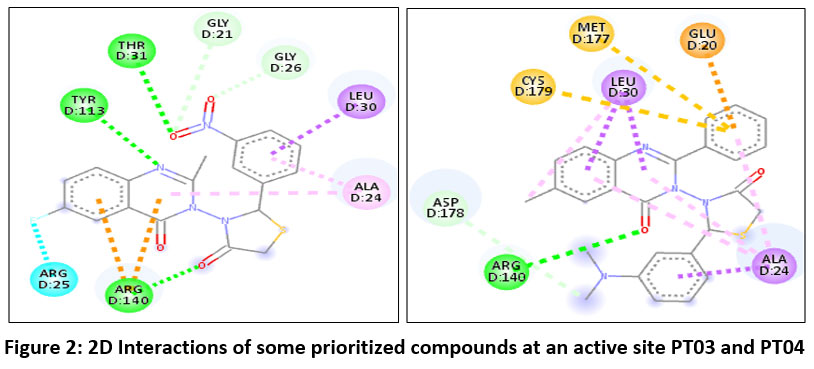 |
Figure 2: 2D Interactions of some prioritized compounds at an active site PT03 and PT04
|
Recent reports (2023–2025) on non-phosphate FBPase inhibitors, particularly indole, oxadiazole, sulfonamide, and heteroaryl-based scaffolds, have demonstrated docking scores typically ranging from −7.5 to −9.5 kcal/mol with key interactions at Arg140, Thr31, and Tyr113 within the AMP/allosteric site. The present quinazolinone–thiazolidinone hybrids, especially PT-04 (−9.7 kcal/mol) and PT-03 (−9.2 kcal/mol), exhibited comparable or superior binding affinity relative to several reported non-phosphonate inhibitors, including N-aryl sulfonyl indole derivatives and diphenyl oxadiazoles. (Figure 2) Importantly, PT-04 established multiple hydrogen bonding interactions closely mimicking the binding pattern observed for established FBPase inhibitors such as CS-917 and MB07803 analogues. 42,43 Furthermore, the absence of a phosphate/phosphonate moiety while retaining strong electrostatic and hydrophobic stabilization suggests improved drug-likeness potential compared to earlier phosphorus-based inhibitors. Collectively, these findings position PT-04 and PT-03 within the affinity range of contemporary non-phosphate FBPase inhibitor candidates under development.24,34,44,45
Chemistry
The synthetic pathway outlined in Figure 3 involves a stepwise heterocyclic ring construction through acylation, cyclization, and carbodiimide-mediated thiazolidinone formation. In the first step, 2-amino-5-fluorobenzoic acid undergoes benzoylation followed by intramolecular cyclodehydration in pyridine to afford 6-fluoro-2-phenyl-4H-benzo[d][1,3] oxazin-4-one (1), representing a classic acylation–cyclization reaction leading to a benzoxazine intermediate. Subsequent treatment of compound (1) with hydrazine hydrate promotes nucleophilic ring opening and rearrangement, yielding 6-fluoro-3-amino-2-phenyl-3,4-dihydroquinazolin-4-one (2) via amino-lysis followed by heterocyclic ring transformation, a well-established route for quinazolinone synthesis. In the final stage, compound (2) reacts with various aromatic or aliphatic aldehydes and thioglycolic acid in the presence of DCC as a coupling agent, facilitating in situ Schiff base formation followed by cyclization to generate the 4-oxothiazolidinone ring. This step represents a one-pot, DCC-mediated cyclo-condensation reaction, efficiently yielding the targeted quinazolinone–thiazolidinone hybrids (PT-01 to PT-10).
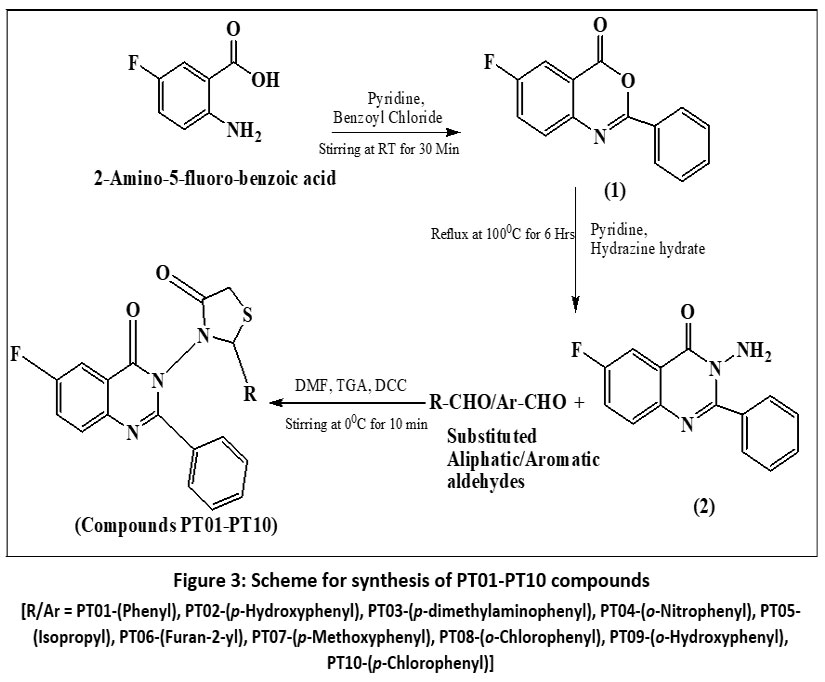 |
Figure 3: Scheme for synthesis of PT01-PT10 compounds
|
Figure 3: [R/Ar = PT01-(Phenyl), PT02-(p-Hydroxyphenyl), PT03-(p-dimethylaminophenyl), PT04-(o-Nitrophenyl), PT05-(Isopropyl), PT06-(Furan-2-yl), PT07-(p-Methoxyphenyl), PT08-(o-Chlorophenyl), PT09-(o-Hydroxyphenyl), PT10-(p-Chlorophenyl)]
The successful formation of the quinazolinone–thiazolidinone hybrid framework in compounds PT-01 to PT-10 was confirmed by IR, 1H NMR, and ESI-MS analyses, with consistent spectral patterns across the series and distinct substituent-dependent variations. In the IR spectra of all compounds, the presence of the quinazolinone core was evidenced by a strong absorption band in the range of 1626–1689 cm⁻¹, corresponding to the amide carbonyl (C=O) stretching vibration. The characteristic C=N stretching of the quinazoline nucleus appeared consistently around 1667–1689 cm⁻¹, while the C–S–C vibration of the thiazolidinone ring was observed between 734–779 cm⁻¹, confirming cyclization. The aryl C–F stretching band was uniformly detected near 1102–1112 cm⁻¹, validating fluorine substitution at the 6-position of the quinazoline ring.
Substituent-specific IR features further supported structural diversity. Hydroxyl-substituted analogues PT-02 and PT-09 exhibited broad absorption bands at 3520 cm⁻¹ and 3327 cm⁻¹, respectively, attributable to phenolic O–H stretching. Nitro-substituted compound PT-04 showed distinct asymmetric and symmetric NO₂ stretching bands at 1524 and 1348 cm⁻¹, while alkyl-substituted PT-05 displayed characteristic alkyl C–H stretching in the 2960–2870 cm⁻¹ region. Halogenated derivatives PT-08 and PT-10 showed additional absorptions around 755–760 cm⁻¹, corresponding to aryl C–Cl stretching.
The 1H NMR spectra of all compounds displayed a multiplet in the range of δ 7.2–8.9 ppm, corresponding to aromatic protons of the quinazoline and phenyl rings, with slight downfield shifts observed for electron-withdrawing substituents such as nitro (PT-04) and chloro (PT-08, PT-10) groups. The thiazolidinone methylene protons (CH₂) appeared as a singlet around δ 3.60–3.75 ppm, while the methine proton (CH) resonated consistently at δ 3.20–3.35 ppm, confirming retention of the heterocyclic ring across the series.
Substituent-specific resonances were clearly identifiable. Compound PT-03 exhibited a singlet at δ ~3.00 ppm corresponding to the dimethyl amino N(CH3)2 protons, while PT-07 showed a singlet at δ ~3.75 ppm due to the methoxy (OCH₃) group. Phenolic protons in PT-02 and PT-09 appeared as singlets at δ 5.30 ppm and δ 5.40 ppm, respectively, indicating free hydroxyl functionality. The isopropyl-substituted analogue PT-05 showed characteristic doublets at δ 1.10–1.25 ppm for methyl groups and a multiplet at δ ~2.9 ppm for the methine proton.
Mass spectrometric analysis further corroborated the proposed molecular structures, with all compounds exhibiting prominent molecular ion peaks ([M]+ or [M+H]+) in agreement with their calculated molecular weights. Halogenated derivatives showed expected isotopic patterns, while protonated molecular ions confirmed structural integrity under ESI conditions. Overall, the spectral data unambiguously confirmed the successful synthesis of the target compounds and demonstrated that electronic and steric variations introduced through different substituents were clearly reflected in their IR and 1H NMR profiles, without perturbing the core quinazolinone–thiazolidinone scaffold.
Biological Evaluation
In-vivo antidiabetic activity
Compounds PT03 was selected for in-vivo hypoglycaemic studies in Swiss albino mice based on the enzyme inhibitory activity and in silico pharmacokinetic predictions. Figure 4 depicts the blood glucose of mice at a 7-day interval during the study. The hypoglycaemic potential of compound PT03 was evaluated in a diabetic animal model and compared with a standard reference antidiabetic drug over a 28-day treatment period. Fasting blood glucose levels were monitored at regular intervals (Days 0, 7, 14, 21, and 28), and the results are summarized in Table 2. No significant changes in body weight, food intake, or water consumption were observed in treatment groups compared to the diabetic control group (p > 0.05). No mortality or abnormal behavioural signs were recorded during the study period, indicating that the synthesized compound was well tolerated at the tested dose levels.
Table 2: Biological activity of Compound PT03
| Days | Group 1 Diabetic Control | Group 2 Treatment with Reference drug | Group 3 Treatment with Compound PT03 at 100 mg/kg | Group 4 Treatment with Compound PT03 at 150 mg/kg |
| 0 | 435 | 522 | 507 | 477 |
| 7 | 475 | 477 | 482 | 433 |
| 14 | 530 | 445 | 452 | 409 |
| 21 | 562 | 422 | 414 | 395 |
| 28 | 584 | 345 | 302 | 382 |
Diabetic Control Group
Animals in the diabetic control group (Group 1) exhibited a progressive and sustained increase in blood glucose levels throughout the study period, rising from 435 mg/dL on day 0 to 584 mg/dL on day 28. This continuous elevation confirms the persistence of hyperglycaemia and validates the suitability of the experimental diabetic model. The absence of glucose reduction also indicates that spontaneous recovery or metabolic adaptation did not occur during the study duration.
Reference Drug Group
Treatment with the reference antidiabetic drug (Group 2) resulted in a marked and time-dependent reduction in blood glucose levels. Glucose levels decreased from 522 mg/dL at day 0 to 345 mg/dL at day 28, representing ~34% reduction. The gradual decline observed from day 7 onward reflects the expected pharmacodynamics response of the standard drug and serves as a benchmark for evaluating the efficacy of PT03. Administration of PT03 at 100 mg/kg (Group 3) produced a pronounced hypoglycaemic effect, with blood glucose levels decreasing from 507 mg/dL on day 0 to 302 mg/dL on day 28, corresponding to an approximate 40% reduction. Notably, the reduction became substantial after day 14, indicating a cumulative or sustained antihyperglycemic effect. By day 28, PT03 at 100 mg/kg demonstrated greater glucose-lowering efficacy than the reference drug, suggesting superior or complementary mechanisms of action. Treatment with PT03 at 150 mg/kg (Group 4) also resulted in a significant reduction in blood glucose levels, decreasing from 477 mg/dL to 382 mg/dL over 28 days (~20% reduction). Although effective, the hypoglycaemic response at this higher dose was less pronounced than that observed at 100 mg/kg, indicating a non-linear dose-response relationship. This observation may reflect saturation of the biological target, altered pharmacokinetics at higher dose, or potential counter-regulatory mechanisms.
Comparative Efficacy and Dose-Response Relationship
Among the treated groups, PT03 at 100 mg/kg emerged as the most effective regimen, achieving the lowest final blood glucose levels and outperforming both the reference drug and the higher PT03 dose. The enhanced efficacy at 100 mg/kg suggests that PT03 possesses an optimal therapeutic window, beyond which increasing the dose does not translate into improved glycaemic control. Such behaviour is consistent with enzyme-targeted mechanisms, including fructose-1, 6-bisphosphatase (FBPase) inhibition, where maximal pathway suppression may occur at intermediate concentrations.
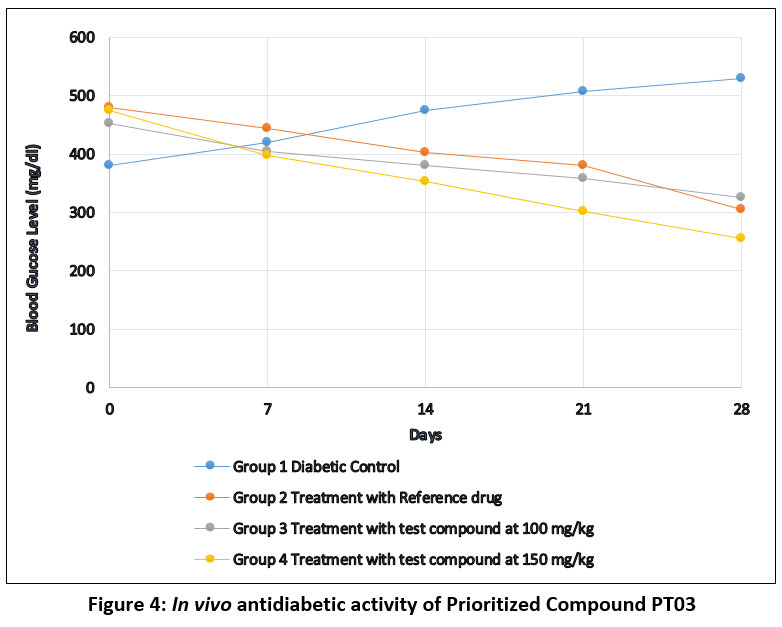 |
Figure 4: In vivo antidiabetic activity of Prioritized Compound PT03
|
Mechanistic Implications
The significant hypoglycaemic activity of PT03 supports its proposed role as an FBPase inhibitor, leading to suppression of hepatic gluconeogenesis and consequent reduction in fasting blood glucose levels. The sustained glucose lowering observed over repeated dosing further suggests favourable metabolic stability and prolonged target engagement.
Discussion
Structure-Activity Relationship (SAR)
Structure–activity relationship analysis of the synthesized quinazoline–thiazolidin-4-one derivatives (PT-01–PT-10) revealed that the nature and position of substituents on the thiazolidinone-linked aromatic moiety significantly influenced binding affinity toward fructose-1,6-bisphosphatase (FBPase) and associated hypoglycaemic activity.
Derivatives bearing unsubstituted or weakly substituted phenyl groups, such as PT-01 (phenyl) and PT-10 (p-chlorophenyl), exhibited moderate docking scores, indicating that hydrophobic interactions alone are insufficient for optimal enzyme inhibition. Introduction of polar hydroxyl substituents improved binding affinity, as observed in PT-02 (p-hydroxyphenyl) and PT-09 (o-hydroxyphenyl), which formed hydrogen-bonding interactions with key residues such as Arg140. However, steric orientation and intramolecular hydrogen bonding appeared to limit further enhancement in activity.
Electron-donating substituents on the aromatic ring led to a pronounced improvement in binding affinity. PT-03, containing a p-dimethyl amino phenyl substituent, showed strong docking interactions and correlated well with its superior in-vivo hypoglycaemic activity. The presence of the dimethyl amino group likely enhances electrostatic complementarity and facilitates multi-point binding within the FBPase pocket. Similarly, PT-07 (p-methoxyphenyl) demonstrated improved docking performance, supporting the beneficial role of electron-rich aromatic systems.
The most significant enhancement in binding affinity was observed for PT-04, bearing a meta-nitrophenyl substituent, which achieved the highest docking score among the series. The electron-withdrawing nitro group promoted multiple hydrogen-bonding and hydrophobic interactions with catalytically relevant residues, resulting in a stable ligand–enzyme complex. In addition, PT-04 displayed favourable in-silico drug-likeness and ADMET properties, further supporting its potential as a lead compound. Aliphatic substitution, as seen in PT-05 (isopropyl), resulted in reduced affinity, underscoring the importance of aromaticity and electronic effects for effective FBPase inhibition.
Overall, the SAR findings indicate that electron-rich or electron-withdrawing aromatic substituents at appropriate positions enhance FBPase binding, while maintaining acceptable drug-like characteristics. These observations provide a rational basis for further optimization of the quinazoline–thiazolidinone scaffold toward the development of effective non-phosphate FBPase inhibitors.
Drug-Likeness and ADMET-Oriented Assessment
The computational drug-likeness evaluation of compounds PT-01–PT-10 was performed using multiple filters, including Lipinski, Ghose, Veber, Egan, and Muegge rules, along with PAINS and Brenk alerts. Most compounds complied with Lipinski’s rule of five, showing at most a single violation, predominantly related to elevated lipophilicity (MLOGP > 4.15) or molar refractivity in a few cases. Importantly, PT-04 fully satisfied all major drug-likeness filters without any violations, reinforcing its suitability as a lead candidate.
Topological polar surface area (TPSA) values ranged from 80.5 to 126.3 Ų, indicating a balance between polarity and membrane permeability, while molecular weights remained within an acceptable range for oral drug development (383–462 Da). Notably, PT-03, despite exhibiting a minor Ghose rule violation due to higher molar refractivity, maintained favourable docking performance and acceptable ADMET descriptors, suggesting that its potent binding may outweigh minor physicochemical liabilities. All compounds were free from PAINS and Brenk structural alerts, indicating low risk of assay interference or reactive toxicity. Collectively, the drug-likeness analysis supports the development potential of this scaffold, with PT-03 and PT-04 emerging as optimal candidates combining strong FBPase binding affinity, favourable interaction profiles, and acceptable drug-like properties.36,46 (Table 1)
The in silico drug-likeness analysis revealed that most compounds complied with Lipinski, Veber, Egan, and Muegge rules, with only minor violations primarily related to lipophilicity or molar refractivity. Importantly, PT-03 and PT-04 showed acceptable molecular weight, TPSA, and hydrogen-bonding characteristics, and all compounds were free from PAINS and Brenk alerts, indicating low risk of assay interference. Considering both computational binding affinity and in-vivo efficacy, PT-03 represents a well-balanced lead candidate for further optimization as a non-phosphate FBPase inhibitor. (Table 3)
Table 3: Drug likeliness prediction
| Comp | MWa | n-rotbb | n-HBDc | n-HBAd | MRe | TPSA
(A⁰) f |
XLOGP3g | WLOGPh | Lipinski | Ghose | Veber | Egan | Muegge |
| PT-01 | 417.46 | 3 | 0 | 4 | 119.25 | 80.5 | 4.41 | 3.83 | 1 | 0 | 0 | 0 | 0 |
| PT-02 | 433.45 | 3 | 1 | 5 | 121.27 | 100.73 | 4.05 | 3.53 | 1 | 0 | 0 | 0 | 0 |
| PT-03 | 460.52 | 4 | 0 | 4 | 133.45 | 83.74 | 4.53 | 3.89 | 0 | 0 | 0 | 0 | 0 |
| PT-04 | 461.45 | 4 | 0 | 6 | 128.07 | 126.32 | 4.23 | 3.74 | 0 | 0 | 0 | 0 | 0 |
| PT-05 | 382.44 | 3 | 0 | 4 | 109.18 | 80.5 | 4.27 | 3.44 | 0 | 1 | 0 | 0 | 0 |
| PT-06 | 407.42 | 3 | 0 | 5 | 111.51 | 93.64 | 3.51 | 3.42 | 0 | 0 | 0 | 0 | 0 |
| PT-07 | 447.48 | 4 | 0 | 5 | 125.74 | 89.73 | 4.38 | 3.84 | 0 | 1 | 0 | 0 | 0 |
| PT-08 | 451.9 | 3 | 0 | 4 | 124.26 | 80.5 | 5.03 | 4.48 | 1 | 1 | 0 | 0 | 0 |
| PT-09 | 433.45 | 3 | 1 | 5 | 121.27 | 100.73 | 4.05 | 3.53 | 1 | 1 | 0 | 0 | 0 |
| PT-10 | 451.90 | 3 | 0 | 4 | 124.26 | 80.5 | 5.03 | 4.48 | 1 | 1 | 0 | 0 | 0 |
Table 3 a: Molecular weight (MW), b: Number of rotatable bonds (n-rotb), c: Number of hydrogen bond donors (n-HBD), d: Number of hydrogen bond acceptors (n-HBA), e: Molecular Refractivity (MR), f: Topological polar surface area (TPSA), g: (XLOGP3), h: (WLOGP)
Toxicological and ADME Evaluation
The In silico ADME and toxicity profiling of the synthesized quinazoline–thiazolidinone derivatives (PT-01–PT-10) was performed to assess their drug-likeness and developability. Overall, the compounds exhibited acceptable absorption, distribution, metabolism, and excretion characteristics, supporting their suitability for further optimization as oral antidiabetic agents. Predicted intestinal permeability parameters, including Caco-2, MDCK, and PAMPA, indicated moderate to good permeability across the series. Aromatic substituted derivatives such as PT-03, PT-04, PT-07, and PT-09 showed comparatively favourable permeability profiles, suggesting efficient passive diffusion. Most compounds were predicted to be non-substrates of P-glycoprotein (P-gp) with minimal efflux liability, indicating a reduced risk of transporter-mediated drug resistance and improved oral bioavailability. Blood–brain barrier (BBB) penetration predictions were within acceptable limits, suggesting limited central nervous system exposure, which is desirable for peripheral antidiabetic therapy.
Cytochrome P450 interaction analysis revealed that the majority of compounds displayed low to moderate inhibitory potential against key CYP isoforms, including CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Notably, PT-03 and PT-04 demonstrated a balanced CYP inhibition profile, minimizing the likelihood of metabolic drug–drug interactions. Predicted human liver microsomal stability and plasma clearance values further supported favourable metabolic behaviour for these compounds.
Toxicological predictions indicated an overall low risk of cardiotoxicity, as reflected by acceptable hERG and hERG-10 µM liability values across the series. All compounds were predicted to be non-mutagenic (AMES-negative) and non-carcinogenic, with low probabilities of hepatotoxicity, nephrotoxicity, neurotoxicity, and skin sensitization. Environmental toxicity parameters, including aquatic toxicity indices, were within permissible limits. Importantly, PT-03, which exhibited the highest docking affinity, also demonstrated a favourable toxicological and ADME profile, complying with drug-likeness filters and FAF-Drugs4 rules. Collectively, the ADME and toxicity evaluation supports the drug-like nature of the synthesized quinazoline–thiazolidinone derivatives. The convergence of favourable pharmacokinetic predictions, low toxicity risk, and strong target binding highlights PT-03 and PT-04 as potential lead candidates for further biological and preclinical investigation as non-phosphate FBPase inhibitors.
The present findings are consistent with previously reported non-phosphate FBPase inhibitors, where heteroaromatic scaffolds such as indole, benzoxazole, and oxadiazole derivatives demonstrated favourable binding within the AMP/allosteric site through interactions with residues including Arg140, Thr31, and Tyr113. Earlier studies have shown that effective inhibition is often associated with multipoint hydrogen bonding combined with hydrophobic stabilization in the adenine-binding pocket, rather than reliance on phosphate mimicry. In this context, the binding profile observed for PT-03 and PT-04, particularly their interactions with Arg140 and surrounding hydrophobic residues (Ala24, Leu30, Met177), aligns with the reported mechanism of action of second-generation non-phosphonate FBPase inhibitors. Furthermore, recent literature emphasizes the importance of optimizing electronic properties and aromatic substitution to enhance electrostatic complementarity and drug-like characteristics, which is reflected in the improved docking performance of electron-donating and electron-withdrawing substituted derivatives in the present series. Thus, the current results support the broader strategy of developing non-phosphate FBPase inhibitors with improved pharmacokinetic potential while maintaining effective enzyme binding.
Conclusion
In the present study, a series of quinazoline–thiazolidin-4-one derivatives were designed and evaluated as potential non-phosphate fructose-1,6-bisphosphatase (FBPase) inhibitors. Molecular docking revealed favourable binding within the adenine-binding pocket, with docking scores ranging from –7.3 to –9.7 kcal/mol. Several derivatives, particularly PT-03 and PT-04, demonstrated stable interactions with key residues through hydrogen bonding and hydrophobic contacts, suggesting appropriate orientation within the enzyme active site.
In the 28-day in-vivo study, PT-03 produced a reduction in blood glucose levels from 507 mg/dL to 302 mg/dL, indicating a measurable anti hyperglycaemic effect. While these findings support the computational predictions and suggest potential modulation of gluconeogenic pathways, the biological evaluation was limited to a small number of compounds and preliminary animal data.
In-silico ADMET and drug-likeness analyses indicated acceptable physicochemical properties for PT-03, including compliance with standard drug-likeness criteria. Collectively, the docking, in-vivo, and computational pharmacokinetic results suggest that the quinazoline-thiazolidinone scaffold warrants further investigation. However, additional mechanistic studies, in-vitro biological screening, and extended in-vivo evaluations are necessary before definitive conclusions regarding potency or therapeutic potential can be established.
Acknowledgement
The authors are profoundly grateful to STES’s Smt. Kashibai Navale College of Pharmacy, Pune for providing facilities to conduct the work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This article does not contain any studies involving human participants performed by any of the authors. This article contains studies involving animal models and prior approval has been taken from Institutional Animal Ethics Committee to carry out the work. (IAEC protocol No: IAEC-176-28/2024) Informed consent was not required for this article. Informed consent was not required for this article.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable
Author Contributions
Vasundhara Sawant: Conceptualization, Analysis, Methodology and Visualization.
Sanjay Sawant, Minal Ghante: Supervision.
Komal Lahoti, Shankar Pawar, Aditi Pawar, Rushikesh Yande, Harshal Waghmare: Data Collection, Writing – Review & Editing, Writing- Original Draft.
References
- McCulloch DK, Campbell IW, Wu FC, Prescott RJ et al. The prevalence of diabetic impotence. 1980;18(4):279–283. doi: 10.1007/BF00288473
CrossRef - Heikkinen S, Argmann CA, Champy MF, Auwerx J. Evaluation of glucose homeostasis. Curr Protoc Mol Biol.2007;77(1):29B.3. doi: 1002/0471142727.mb29b03s77
CrossRef - Lorenzati B, Zucco C, Miglietta S, Lamberti F. Oral hypoglycemic drugs: Pathophysiological basis of their mechanism of action. 2010;3(9):3005–3020. doi: 10.3390/ph3093005
CrossRef - IDF 1 Sun H, Saeedi P, Karuranga S, Pinkepank M, et.al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes res. Clinic. Pract..2022;183(1):109-119. doi: 1186/s12916-024-03401-3.
CrossRef - Kumar A, Gangwar R, Ahmad Zargar A, et.al. Prevalence of diabetes in India: A review of IDF diabetes atlas. Curr Diabetes Rev, 2024;20(1):e130423215752. doi: https://doi.org/10.2174/1573399819666230413094200
CrossRef - Moller DE. New drug targets for type 2 diabetes and the metabolic syndrome. 2001,414: 821–827. doi: 10.1038/414821a
CrossRef - Botton G, Leriche C, De Vacqueur A. Benzimidazole dihydro-thiadiazinone derivatives as fructose-1,6-bisphosphatase inhibitors and pharmaceutical compositions comprising same. US Patent US8232272B2; 2012.
- Rendell MS. Current and emerging gluconeogenesis inhibitors for the treatment of type 2 diabetes. Expert Opin Pharmacother. 2021;22(16):2167–2179. doi: 1080/14656566.2021.1975095
CrossRef - Mehanna A. Antidiabetic agents: Past, present and future. Future Med Chem. 2013;5(4):411–430. doi: 4155/fmc.13.17
CrossRef - Madiraju AK, Erion DM, Rahimi Y. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. 2014;510:542–546. doi: 10.1038/nature13270
CrossRef - Krishnan B, Babu S, Walker J, Walker AB et.al. Gastrointestinal complications of diabetes mellitus. World J Diabetes. 2013;4(3):51–63. doi: 4239/wjd.v4.i3.51
CrossRef - Cao C, Chen YW, Wu Y, Deumens E. Opal: A multiscale multi center simulation package based on MPI-2 protocol. Int J Quantum Chem. 2011;111:4020–4029. doi: 1002/qua.22672
CrossRef - Sargent MV, Stransky PO, Carney JR, Krenitsky JM, et al. Dibenzofuran derivatives as inhibitors of fructose-1,6-bisphosphatase and methods of use thereof. US Patent US9273021B2.
- Gizak A, Duda P, Wiśniewski J, Rakus D. Fructose-1,6-bisphosphatase: From glucose metabolism enzyme to multifaceted regulator of cell fate. Adv Biol Regul.2019;72:41–50. doi: 10.1016/j.jbior.2019.02.003
CrossRef - Xu Y, Huang Y, Song R. Development of disulfide-derived fructose-1,6-bisphosphatase covalent inhibitors for treatment of type 2 diabetes. Eur J Med Chem. 2020;203:112500. doi: 1016/j.ejmech.2020.112500
CrossRef - Silva LCL, de Souza GH, Ames-Sibin AP, et al. Inhibition of gluconeogenesis by boldine in the perfused liver: therapeutical implication for glycemic control. Int J Hepatol, 2023;1:1283716. doi: 10.1155/2023/1283716
CrossRef - Barciszewski J, Szpotkowski K, Wiśniewski J. Structural studies of human muscle FBPase. Acta Biochim Pol. 2021;68(1):5-14. doi: 18388/abp.2020_5473
CrossRef - Baig MS, Suryawanshi RM, Zehravi M. Surface decorated quantum dots: Synthesis, properties and role in herbal therapy. Front Cell Dev Biol. 2023;11:1139671. doi: 3389/fcell.2023.1139671
CrossRef - Banu S, Bhowmick A. Therapeutic targets of type 2 diabetes: An overview. Moj Drug Des Dev Ther. 2017;1(3):60–64.
CrossRef - Belete TM. Recent achievements in discovery and development of novel targets for type 2 diabetes mellitus. J Exp Pharmacol. 2020;12:1–15. doi: 2147/JEP.S224965
CrossRef - Pernicova I, Korbonits M. Metformin-Mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10(3):143-156. doi: 1038/nrendo.2013.256
CrossRef - Hunter RW, Hughey CC, Lantier L. Metformin reduces liver glucose production by inhibition of fructose-1,6-bisphosphatase. Nat Med. 2018;24:1395-1406. doi: 1038/s41591-018-0159-7
CrossRef - Gormsen LC, Søndergaard E, Christensen NL, Brøsen K, et al. Metformin increases endogenous glucose production in non-diabetic individuals and individuals with recent-onset type 2 diabetes. 2019;62(7):1251-1256. doi: 10.1007/s00125-019-4887-7
CrossRef - Zhou J, Bie J, Wang X. Discovery of N-arylsulfonyl-indole-2-carboxamide derivatives as potent fructose-1,6-bisphosphatase inhibitors. J Med Chem. 2020;63(18):10307–10329. doi: 10.1021/acs.jmedchem.0c01092
CrossRef - Zhang X, Yang S, Chen J, Su Z. Regulation of hepatic gluconeogenesis. Front Endocrinol. 2019;(10):802. doi: 3389/fendo.2019.00802
CrossRef - Balasopoulou A, Kokkinos P, Pagoulatos D. Recent advances and challenges in management of retinoblastoma. BMC Ophthalmol. 2017;(17):1. doi: 1186/s12886-017-0523-3
CrossRef - Lai C, Gum RJ, Daly M. Benzoxazole benzene sulfonamides as allosteric inhibitors of fructose-1,6-bisphosphatase. Bioorg Med Chem Lett. 2006;(16):1807–1810. doi: 1016/j.bmcl.2006.02.051
CrossRef - Von Geldern TW, Lai C, Gum RJ. Benzoxazole benzene sulfonamides as novel allosteric inhibitors of fructose-1,6-bisphosphatase. Bioorg Med Chem Lett. 2006;(16):1811–1815. doi: 1016/j.bmcl.2006.02.050
CrossRef - Bie J, Liu S, Li Z, Mu Y, Xu B, Shen Z. Discovery of novel indole derivatives as allosteric inhibitors of fructose-1,6-bisphosphatase. Eur J Med Chem. 2015;(90):394–405. doi: 10.1016/j.ejmech.2015.02.018
CrossRef - Ligand Pharmaceuticals. Safety, tolerability and efficacy study of MB07803 administered to patients with type 2 diabetes mellitus. ClinicalTrials.gov Identifier: NCT00458016.
- Van Poelje PD, Potter SC, Fujitaki JM, Dang Q, et al. MB07803, a prodrug of second-generation FBPase inhibitor, lowers blood glucose in diabetic models. 2008;57(1):A101.
- Ali, Zulphikar, Akhtar, Md Jawaid, Haider et. al. Design and synthesis of quinazoline-3,4-(4H)-diamine endowed with thiazoline moiety as new class for DPP-4 and DPPH inhibitor. Bioorganic Chem. 2017;71(1):181-191. doi: 1016/j.bioorg.2017.02.004
CrossRef - Khan S, Bjij I, Soliman MES. Selective covalent inhibition of allosteric Cys121 distorts binding of PTP1B enzyme. Cell Biochem Biophys. 2019;77(3):203–211. doi: 1007/s12013-019-00869-8
CrossRef - Desai, N.C., Jadeja, K.A., Jadeja, et.al. Design, synthesis, antimicrobial evaluation, and molecular docking study of some 4-thiazolidinone derivatives containing pyridine and quinazoline moiety. Communications. 2021;51(6):952-963. doi: 10.1080/00397911.2020.1861302
CrossRef - Kitas E, Mohr P, Kuhn B. Sulfonylureido thiazoles as fructose-1,6-bisphosphatase inhibitors for treatment of type 2 diabetes. Bioorg Med Chem Lett. 2010;20(2):594–599. doi: 10.1016/j.bmcl.2009.11.126
CrossRef - Liao BR, He HB, Yang LL. Non-phosphorus-based fructose-1,6-bisphosphatase inhibitors: SAR of diphenyl oxadiazoles. Eur J Med Chem. 2014;(83):15–25. doi: 10.1016/j.ejmech.2014.06.041
CrossRef - Ke H, Zhang Y, Lipscomb WN. Crystal structure of fructose-1,6-bisphosphatase complexed with fructose-6-phosphate, Amp and magnesium. Proc Natl Acad Sci USA. 1990;87(14):5243–5247. doi: 1073/pnas.87.14.5243
CrossRef - Sampat G, Suryawanshi SS, Palled MS, et al. Drug likeness screening and physicochemical evaluation using Cadd tools. Adv Pharmacol Pharm. 2022;10(4):234–246. doi: 10.13189/app.2022.100402
CrossRef - Fu L, Shi S, Yi J, Wang N, He Y, et al. Advances in nucleic acid research tools. Nucleic Acids Res. 2024;(1): gkae236. doi: 1093/nar/gkae236
CrossRef - Miao M, Niu Y, Xie T, Yuan B, et al. Diabetes-impaired wound healing and macrophage activation. Wound Repair Regen. 2012;20(2):203–213. doi: 10.1111/j.1524-475X.2012.00783.x
CrossRef - Sawant SD, Sawant VN. Comprehensive review and perspective on fructose 1,6-bisphosphatase inhibitors for the management of type 2 diabetes mellitus. Russ J Bioorg Chem. 2025;(51):439–464. doi: 1134/S1068162025050142
CrossRef - Walker, JR, Triscari, J Freudenthaler. Pharmacokinetics and Pharmacodynamics of Single-Dose CS-917, a Novel Prodrug of a Fructose 1, 6-Bisphosphatase Inhibitor, in Patients with Type 2 Diabetes. Diabetes. 2007;1(1):54-56. doi: 1073/pnas.0502983102
CrossRef - Van Poelje, P. D., Potter, S. C., Fujitaki. MB07803, a Prodrug of a Second Generation Fructose-1,6-Bisphosphatase Inhibitor, Lowers Blood Glucose in Diabetic Rodents and in Cynomolgus Monkeys. Diabetes. 2008;57(1), A101-A110. doi: https://doi.org/10.1039/9781849734141-00306
- Sawant V, Sawant S, Tamte P, Ghante M, Tangde J. Exploring the multi-targeted therapeutic mechanism of bioactives from Glycine max in treatment of type 2 diabetes mellitus and obesity through an in-vitro and in-silico approach. Biotechnol Res Asia. 2025;22(2),637-648. doi: 10.13005/bbra/3390
CrossRef - Wang, Xiaoyu Zhao, Rui Ji, et. al. Discovery of Novel Indole Derivatives as Fructose-1,6-bisphosphatase Inhibitors and X-ray Cocrystal Structures Analysis, ACS Med. Chem. Letters, 2022;13(1), 118-127. doi: 10.1021/acsmedchemlett.1c0061
CrossRef - Sampat G, Suryawanshi SS, Palled MS et. al. Drug likeness screening and evaluation of physicochemical properties of selected medicinal agents by computer aided drug design tools. Pharmacol. Pharm, 2022;10(4), 234-246. doi: 10.13189/app.2022.100402
CrossRef
Abbreviations
FBPase – Fructose-1,6-bisphosphatase, T2DM – Type-2 Diabetes Mellitus, SAR – Structure–Activity Relationship, ADMET – Absorption, Distribution, Metabolism, Excretion and Toxicity, HBD – Hydrogen Bond Donor, HBA – Hydrogen Bond Acceptor, MW – Molecular Weight, LogP – Partition Coefficient, TPSA – Topological Polar Surface Area, RMSD – Root Mean Square Deviation, IR – Infrared Spectroscopy, ¹H NMR – Proton Nuclear Magnetic Resonance, MS – Mass Spectrometry, PDB – Protein Data Bank, PK – Pharmacokinetics, RCSB: Research Collaboratory for Structural Bioinformatics; BBB: Blood Brain Barrier
Accepted on: 08-03-2026
Second Review by: Dr. Nawaf A. Almolhis
Final Approval by: Dr. Eugene A. Silow








![]()
![]()
