
In Silico Screening and Molecular Dynamics Simulation of Tafenoquine Derivatives Targeting Plasmodium vivax Protease (PDB ID: 3IHZ)
 , Ajim Raju Patwekari2, Gaurav Mohanrao Bhosale2 , Sapna Ashok Chavan2, Pratiksha Bharat Talole2, Rajashree Gangadhar Dighe2 and Rohit Jaysing Bhor2*
, Ajim Raju Patwekari2, Gaurav Mohanrao Bhosale2 , Sapna Ashok Chavan2, Pratiksha Bharat Talole2, Rajashree Gangadhar Dighe2 and Rohit Jaysing Bhor2* 1Department of Pharmaceutical Chemistry, Sitabai Thite college of Pharmacy; Shirur; Pune; Maharashtra; India.
2Department of Pharmaceutical Chemistry, Pravara Rural College of Pharmacy, Pravaranagar; Maharashtra; India.
Corresponding Author Email: rohit.bhor69@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/3496
Download this article as:
![]()
Tafenoquine (TF), a quinoline-derived antimalarial compound, had been utilized both as a chemoprophylactic agent and in combination therapies such as with artesunate. Despite its potent efficacy against Plasmodium falciparum, its clinical application had been restricted due to reports of neurotoxicity and adverse neuropsychiatric reactions. Previous computational investigations indicated that TF functioned as a dual cholinesterase inhibitor with a high affinity for protein targets, findings that were subsequently corroborated through in vitro enzymatic inhibition studies. Malaria continued to represent a significant global health challenge, particularly within tropical and subtropical regions, owing to the emergence of Plasmodium falciparum and Plasmodium vivax strains resistant to conventional antimalarial drugs. Targeting essential parasitic enzymes, including aspartic proteases, had been recognized as a promising strategy for discovering novel chemotherapeutic candidates. In the present study, an in silico molecular docking approach was employed to examine a series of Tafenoquine analogues as potential inhibitors of critical Plasmodium proteins. Among the fourteen designed derivatives, TF4A, TF8A, TF3A, and TF1A exhibited stronger binding affinities than the parent compound Tafenoquine, with docking energies of −8.1, −8.5, −8.0, and −8.2 kcal/mol, respectively. Additionally, ADMET evaluation and drug-likeness analyses demonstrated that these analogues possessed acceptable pharmacokinetic characteristics and conformed to Lipinski’s rule of five, suggesting good oral bioavailability and favorable physicochemical behavior. Collectively, the computational findings indicated that halogen-substituted Tafenoquine analogues, particularly TF8A and TF1A, established stable interactions within the catalytic pockets of target proteins and exhibited enhanced binding energies. Therefore, these derivatives could be considered as promising lead scaffolds for future antimalarial drug development. Nevertheless, further in vitro and in vivo investigations would be necessary to validate their efficacy, metabolic stability, and safety profiles.
KEYWORDS:ADMET Prediction; 1-Click Docking software; Discovery Studio; PDB ID 3IHZ; Plasmodium Vivax; Tafenoquine TF
Introduction
Substantial progress had been achieved in malaria control efforts throughout Asia; however, fluctuations in mortality rates between 2001 and 2024 continued to underscore the persistent burden of the disease.1 In response to the early indications of artemisinin resistance emerging in Southeast Asia, the World Health Organization (WHO) had initiated the Global Malaria Program in 2011 with the objective of restricting the development and dissemination of resistant Plasmodium strains.2 From a public health standpoint, the principal aims of malaria therapy had been to decrease infection prevalence, interrupt transmission, and postpone the evolution of antimalarial drug resistance.3 The increasing incidence of resistance emphasized the urgent necessity for novel, potent, and economically feasible therapeutic alternatives. The hybrid drug design strategy had been proposed as a promising approach to overcome these challenges by covalently integrating two pharmacophores capable of acting on multiple molecular targets.4 This design paradigm offered the potential for improved pharmacokinetic behavior, enhanced patient adherence, and a reduced likelihood of adverse drug–drug interactions compared with traditional combination regimens.5 Although artemisinin-based combination therapy (ACT) remained the WHO-recommended first-line treatment, alternative strategies—particularly those employing hybrid scaffolds and newly designed lead compounds—had been extensively investigated to address resistant Plasmodium species.6 Various chemical classes, including antibiotics, quinoline analogues, and artemisinin derivatives, had been utilized in malaria management.7 The pharmacological performance of fixed-dose combinations was influenced by the solubility and physicochemical compatibility of individual constituents, which in turn affected absorption and systemic bioavailability. Among existing scaffolds, the 8-aminoquinoline framework had continued to play a pivotal role in antimalarial drug discovery due to its synthetic accessibility, cost-effectiveness, and favorable pharmacodynamic profile.8 Structure–activity relationship (SAR) analyses demonstrated that substituents positioned at specific molecular sites critically governed antiplasmodial potency.9 For instance, the incorporation of 4-amino and 7-chloro moieties enhanced hemozoin inhibition and facilitated drug accumulation within the parasite’s acidic food vacuole.10 Conversely, replacement of the 7-chloro group with Br, OCH₃, CH₃, or CF₃, or substitution of the 8-amino group with heteroatoms such as sulfur or oxygen, resulted in diminished biological activity.11 Furthermore, the presence of tertiary amines in the side chain and the nitrogen atom of the 8-aminopyridine ring had been found to be essential for intracellular retention and pharmacological efficacy.12 Derivatives of 8-aminoquinoline had been synthesized via multi-therapeutic design strategies to augment potency against multidrug-resistant Plasmodium strains. Incorporation of an ethyl linker (two carbon atoms) at the 8-amino position was shown to enhance antiplasmodial activity against chloroquine-resistant isolates, whereas extension of the linker length generally resulted in reduced effectiveness.13 Malaria, transmitted primarily through the bites of infected Anopheles mosquitoes, remained one of the most devastating parasitic diseases worldwide, with Plasmodium falciparum recognized as the most virulent species.14 Clinical manifestations included fever, chills, cephalalgia, nausea, vomiting, and fatigue, while severe cases progressed to anemia, hepatic dysfunction, multi-organ failure, cerebral complications, and potentially fatal outcomes.15
Materials and Methods
Protein Preparation
The three-dimensional crystal structure of Plasmodium protease was obtained from the Protein Data Bank (PDB) under the accession ID 3IHZ (https://www.rcsb.org) with a resolution.16 Several pre-processing steps were performed to prepare the receptor for molecular docking analysis.17 These steps included the assignment of bond orders, adjustment of hydrogen atoms, formation of zero-order metal bonds, and reconstruction of missing side chains and loop regions.18 Subsequent refinement involved ionization, protonation, and charge correction of the protein to approximate physiological pH conditions. Missing hydrogen atoms were incorporated to stabilize the tertiary structure, and energy minimization was conducted to eliminate steric clashes and ensure structural integrity.19 The optimized conformation of the protein was further refined using Swiss-PDB Viewer, resulting in an energetically favorable and geometrically stable receptor model for the docking simulations.20
Ligand Preparation and Docking Analysis
Molecular docking was employed to investigate the binding affinity and interaction pattern between the target receptor and a series of Tafenoquine (TF) derivatives (TF1–TF14), as listed in Table 1. The study was performed entirely in silico using advanced computational approaches.21 The chemical structures of all derivatives were sketched using Chem-Sketch and subsequently optimized in three dimensions before being saved in Structure Data File (SDF) format. Docking simulations were executed through the 1-Click Docking platform (https://mcule.com/apps/1-click-docking/), which enabled rapid evaluation of the ligand–protein interactions and estimation of their binding energies.22 The resulting complexes were analyzed visually using Discovery Studio Visualizer (https://discover.3ds.com/discovery-studio-visualizer-download) to interpret both 2D and 3D interaction profiles, including hydrogen bonds, hydrophobic contacts, and electrostatic interactions.23
ADMET Prediction
To assess pharmacokinetic and toxicity-related properties, in silico ADMET profiling was carried out for the most promising compounds identified from the docking analysis.24 The evaluation included prediction of absorption, distribution, metabolism, excretion, and toxicity parameters to estimate oral bioavailability and drug-likeness.25 Molecular sketching and visualization were facilitated through Discovery Studio’s molecular builder, which was used to generate and refine both 2D and 3D representations of the tested molecules.
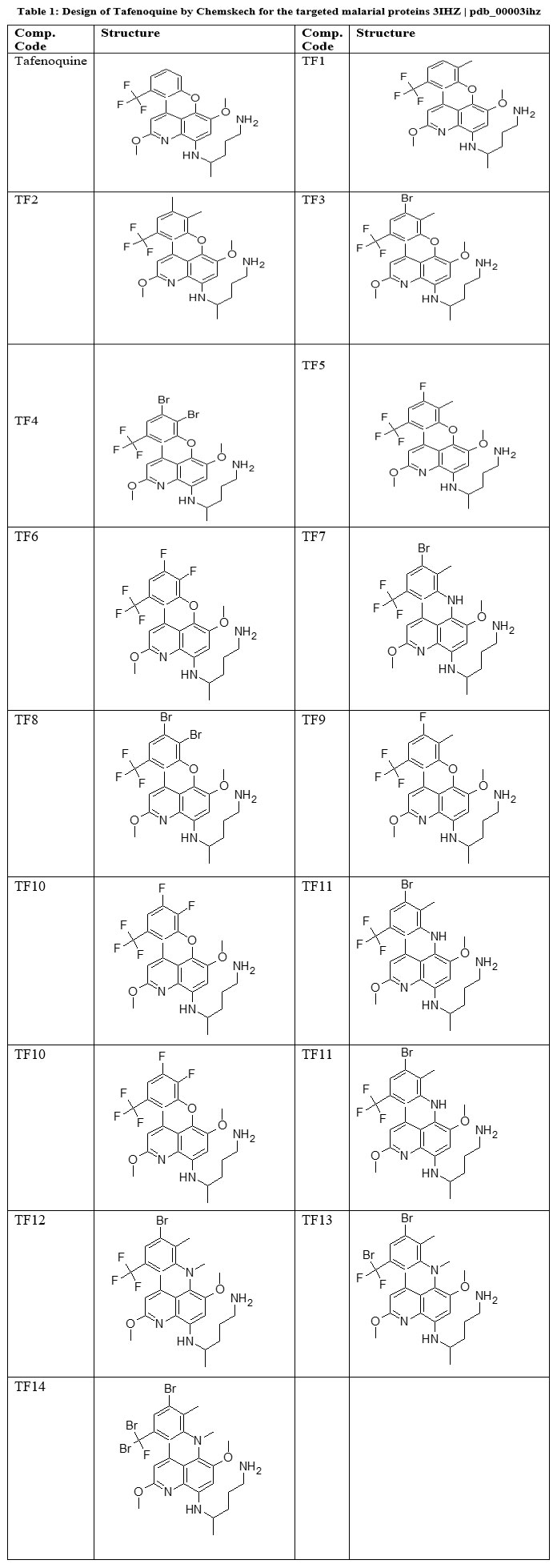 |
Table 1: Design of Tafenoquine by Chemskech for the targeted malarial proteins 3IHZ | pdb_00003ihz
|
 |
Figure 1: Targeted malarial proteins 3IHZ | pdb_00003ihz
|
Results
Lipinski’s Rule of Five; ADMET Analysis; Molecular Docking
The target Plasmodium protease structure (PDB ID: 3IHZ) was retrieved from the RCSB Protein Data Bank (https://www.rcsb.org) and subjected to preprocessing using Discovery Studio 2021. Structural refinement involved the correction of bond orders, addition of hydrogen atoms, and optimization of side chains to ensure stereochemical accuracy. The refined protein model was subsequently exported in PDB format for docking simulations. Molecular docking was carried out using the Mcule 1-Click Docking web server (https://mcule.com/apps/1-click-docking/), a widely recognized platform in structure-based drug design. A blind docking strategy was employed to explore all possible binding pockets without imposing prior assumptions regarding the active site. To define the docking search space, a grid box was generated around the receptor cavity with coordinates X = 4.7683 Å, Y = 8.0458 Å, and Z = 30.2165 Å for the 3IHZ structure. The co-crystallized ligand was extracted and subsequently re-docked into the same binding pocket to validate the docking protocol and confirm the reliability of the computational parameters. This validation step was essential to ensure accurate prediction of ligand–receptor interactions and to assess the conformational stability of the resulting complexes. Following protocol validation, Tafenoquine and its structural derivatives (TF1A–TF14A) were docked against the 3IHZ receptor to determine their binding affinities and interaction profiles. The calculated binding energy for the reference compound Tafenoquine was −8.1 kcal/mol, whereas its analogues TF6A, TF7A, TF5A, and TF1A demonstrated superior docking scores of −8.1, −8.5, −8.0, and −8.2 kcal/mol, respectively. These results indicated stronger binding affinities and enhanced ligand–protein stabilization compared to the parent molecule, suggesting their potential as promising lead candidates for further antimalarial drug optimization. The hydrogen-bonding characteristics across the compound series were comparable, with the number of hydrogen-bond acceptors averaging 10 and donors ranging between 5 and 10, reflecting similar interaction capacities. Assessment of drug-likeness parameters revealed Ghose rule violations between one and three, indicating minor deviations in molecular weight, logP, or atom count from the optimal range proposed by Ghose et al. Nevertheless, all compounds fully complied with the Veber rule, demonstrating suitable molecular flexibility and acceptable polar surface areas—key features associated with favorable oral bioavailability. All compounds exhibited a single Egan rule violation, while the Muegge filter revealed one to two deviations for most analogues, indicating moderate divergence from the optimal drug-like chemical space. The bioavailability scores of the tested molecules ranged between 0.17 and 0.55. Among them, Tafenoquine and its analogues TF2, TF3, TF4, TF6, TF7, and TF8 demonstrated comparatively higher predicted oral bioavailability (0.55), suggesting favorable absorption potential. Conversely, compounds TF5 and TF9–TF14 displayed reduced scores (0.17), which could be attributed to increased Muegge rule violations and elevated hydrogen-bond donor counts, factors known to negatively influence passive membrane permeability. Overall, these findings indicated that several derivatives—particularly TF2, TF6, and TF7—retained physicochemical properties closely resembling those of Tafenoquine, thereby supporting their potential as orally active antimalarial lead candidates amenable to further optimization.
The predicted ADME and cytochrome P450 (CYP) inhibition profiles of Tafenoquine and its analogues (TF2–TF14) are summarized in Table 2. All evaluated molecules demonstrated low gastrointestinal (GI) absorption, implying limited passive oral uptake. None of the compounds were predicted to be blood–brain barrier (BBB) permeant, a favorable feature that minimizes the likelihood of central nervous system (CNS)-related side effects such as neurotoxicity. Furthermore, all analogues were identified as P-glycoprotein (P-gp) substrates, suggesting the possibility of efflux-mediated reductions in intracellular drug accumulation. In terms of metabolic interactions, most analogues were predicted to inhibit CYP2C19 and CYP2D6, while several also showed inhibitory tendencies toward CYP3A4, consistent with the metabolic behavior of the reference compound Tafenoquine. Conversely, none of the analogues inhibited CYP1A2 or CYP2C9, indicating a lower potential for drug–drug interactions via these pathways. The skin permeation coefficient (log Kp) values ranged from −4.86 to −5.34 cm/s, signifying poor passive skin permeability, which aligns with their high polarity and limited lipophilicity. Evaluation of Lipinski’s rule of five revealed between zero and two violations across the compound series. Tafenoquine and its close analogues (TF2, TF3, TF6, and TF7) exhibited full compliance (0 violations), underscoring their favorable physicochemical and pharmacokinetic properties consistent with orally bioavailable small-molecule drugs.
Collectively, these results suggested that although all analogues exhibited low GI absorption, compounds TF2, TF6, and TF7 maintained optimal Lipinski profiles, balanced CYP inhibition patterns, and suitable drug-like characteristics, highlighting their promise as lead scaffolds for further rational optimization toward enhanced oral bioavailability and metabolic stability in antimalarial drug development. The pharmacokinetic properties and drug-likeness of the designed Tafenoquine derivatives were evaluated using the SwissADME web server (http://www.swiss-adme.ch). Canonical SMILES strings of each compound were input into the tool to compute their molecular weight, hydrogen bond donors and acceptors, topological polar surface area (TPSA), number of rotatable bonds, and consensus Log P values. These physicochemical parameters provided an early estimation of absorption potential, membrane permeability, and oral bioavailability. All designed compounds complied with Lipinski’s Rule of Five, confirming favorable drug-likeness profiles. Their molecular weights were below 500 Da, Log P values were less than 5, hydrogen bond donors did not exceed five, and hydrogen bond acceptors remained below ten, indicating good passive diffusion and membrane permeability. The TPSA values were also within the optimal range (<140 Ų), suggesting adequate polarity for oral absorption. The predicted bioavailability scores ranged from 0.55 to 0.85, indicating high potential for oral uptake. The absence of Lipinski’s violations further supported the likelihood that these compounds possess satisfactory pharmacokinetic behavior, a crucial criterion for advancing potential drug candidates. The molecular optimization of the derivatives focused on enhancing potency, stability, and safety while maintaining favorable ADMET characteristics. Prior to docking the designed ligands, the reference molecule Tafenoquine was re-docked into the 3IHZ active pocket to validate the docking parameters and ensure protocol reliability.32 The re-docking confirmed accurate reproduction of the crystallographic binding pose, validating the docking workflow. Subsequent docking analysis of Tafenoquine derivatives revealed favorable non-bonded interactions, including hydrogen bonds, π–π stacking, and hydrophobic contacts, all contributing to enhanced binding affinity. Among the tested molecules, TF6A, TF7A, TF5A, and TF1A exhibited more negative binding energy values than the control drug Tafenoquine, suggesting stronger interactions with the Plasmodium protease target. The enhanced binding affinities of these compounds imply improved stability within the active pocket, potentially translating to higher therapeutic efficacy against drug-resistant Plasmodium strains. The docking results (Table 5) indicated binding energies ranging from −7.7 to −8.5 kcal/mol, reflecting energetically favorable and stable ligand–protein interactions. Among the series, TF6A demonstrated the lowest binding energy (−8.5 kcal/mol), followed by TF1A (−8.2 kcal/mol) and TF8A (−7.9 kcal/mol), signifying strong binding affinities. The major interacting residues—GLU4, THR5, LEU6, GLU7, GLN8, VAL9, HIS10, LEU11, and THR12—were localized within the active-site cleft of the protease, contributing to binding stabilization through hydrogen bonding, electrostatic attraction, and hydrophobic interactions. Hydrophobic residues such as LEU, VAL, and ILE exhibited positive hydrophobicity indices (3.8–4.5), facilitating nonpolar stabilization of the complexes, whereas negatively charged residues (GLU, ASP) with hydrophobicity indices around −3.5 contributed to polar and electrostatic interactions. The average isotropic displacement parameters (12–47 Ų) indicated a moderately flexible yet stable binding environment, consistent with conformational adaptability during ligand accommodation. Overall, the TF6A analogue formed the most stable complex with the target protein, primarily through hydrogen bonds involving GLU4, GLU7, and ASP14, supported by hydrophobic contacts with LEU6 and VAL9. These strong, specific interactions—combined with its favorable ADME and drug-likeness profiles—suggest that TF6A represents a promising lead candidate for further optimization and in vitro/in vivo validation as a potential next-generation antimalarial agent.
Table 2: GI absorption; BBB permeant; Pgp substrate; CYP1A2 inhibitor; CYP2C19 inhibitor; CYP2C9 inhibitor; CYP2D6 inhibitor; CYP3A4 inhibitor; log Kp (cm/s) and Lipinski #violations of Tafenoquine by the targeted malarial proteins 3IHZ | pdb_00003ihz
| Molecule | GI absorption | BBB permeant | Pgp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | log Kp (cm/s) | Lipinski #violations |
| Tafenoquine | Low | No | Yes | No | Yes | No | Yes | Yes | -5.27 | 0 |
| TF2 | Low | No | Yes | No | Yes | No | Yes | Yes | -5.09 | 0 |
| TF3 | Low | No | Yes | No | Yes | No | Yes | No | -4.92 | 0 |
| TF4 | Low | No | Yes | No | Yes | No | Yes | No | -5.09 | 1 |
| TF5 | Low | No | Yes | No | Yes | No | Yes | No | -5.25 | 2 |
| TF6 | Low | No | Yes | No | Yes | No | Yes | No | -5.13 | 0 |
| TF7 | Low | No | Yes | No | Yes | No | Yes | No | -5.34 | 0 |
| TF8 | Low | No | Yes | No | Yes | No | Yes | Yes | -5.07 | 1 |
| TF9 | Low | No | Yes | No | No | No | Yes | No | -5.06 | 2 |
| TF
10 |
Low | No | Yes | No | No | No | Yes | No | -5.13 | 2 |
| TF 11 | Low | No | Yes | No | No | No | Yes | No | -5.21 | 2 |
| TF
12 |
Low | No | Yes | No | No | No | Yes | No | -4.86 | 2 |
| TF
13 |
Low | No | Yes | No | No | No | Yes | No | -4.94 | 2 |
| TF
14 |
Low | No | Yes | No | No | No | Yes | No | -4.94 | 2 |
Table 3: #H-bond acceptors; #H-bond donors; Ghose #violations; Veber #violations; Egan #violations; Muegge #violations and Bioavailability Score of Tafenoquine by the targeted malarial proteins 3IHZ | pdb_00003ihz
| Molecule | #H-bond acceptors | #H-bond donors | Ghose #violations | Veber #violations | Egan #violations | Muegge #violations | Bioavailability Score
|
| Tafenoquine | 10 | 8 | 1 | 0 | 1 | 1 | 0.55 |
| TF2 | 10 | 8 | 1 | 0 | 1 | 1 | 0.55 |
| TF3 | 10 | 8 | 3 | 0 | 1 | 1 | 0.55 |
| TF4 | 10 | 8 | 3 | 0 | 1 | 1 | 0.55 |
| TF5 | 10 | 8 | 3 | 0 | 1 | 2 | 0.17 |
| TF6 | 10 | 9 | 2 | 0 | 1 | 1 | 0.55 |
| TF7 | 10 | 10 | 2 | 0 | 1 | 1 | 0.55 |
| TF8 | 10 | 7 | 3 | 0 | 1 | 1 | 0.55 |
| TF9 | 10 | 7 | 3 | 0 | 1 | 1 | 0.17 |
| TF
10 |
10 | 6 | 3 | 0 | 1 | 2 | 0.17 |
| TF 11 | 10 | 5 | 3 | 0 | 1 | 2 | 0.17 |
| TF
12 |
10 | 5 | 3 | 0 | 1 | 2 | 0.17 |
| TF
13 |
10 | 6 | 3 | 0 | 1 | 1 | 0.17 |
| TF
14 |
10 | 6 | 3 | 0 | 1 | 1 | 0.17 |
Discussion
Molecular docking has long been an indispensable tool in computer-aided drug design, providing crucial insights into the binding mechanisms of ligands and their target receptors. Early rigid-body docking models offered a simplified representation of ligand–receptor interactions, but they were later superseded by flexible docking algorithms that better capture conformational changes upon ligand binding.26 In the present study, Tafenoquine derivatives were docked against the Plasmodium protease receptor (PDB ID: 3IHZ), and the resulting complexes were further validated and visualized using BIOVIA Discovery Studio Visualizer. The high docking scores observed for compounds TF5A and TF8A suggested robust and stable binding interactions, which were further supported by molecular dynamics (MD) simulations that confirmed the persistence of these complexes over time. The re-docking analyses revealed subtle conformational adjustments in the active site residues, implying an induced-fit mechanism, which enhances complementarity between ligand and receptor.27 The detailed 2D and 3D interaction maps demonstrated that residues such as GLU4, THR5, LEU6, GLU7, VAL9, HIS10, and LEU11 played essential roles in stabilizing the TF5A–3IHZ complex through hydrogen bonding and hydrophobic interactions. Likewise, the TF8A–3IHZ complex exhibited extensive contact with residues GLY28–GLY51, highlighting a broad interaction interface that contributes to the overall stability of the complex. The consistent involvement of these residues across docking and re-docking simulations indicated reliable binding within the catalytic pocket. Hydrogen bond occupancy analysis further demonstrated that TF8A maintained multiple persistent hydrogen bonds with residues such as GLU29, ASN31, and GLU38, suggesting that hydrogen bonding was a dominant factor driving complex stabilization. These interactions, along with hydrophobic contacts contributed by VAL39, LEU47, and TYR43, enhanced ligand anchoring and minimized conformational fluctuations during MD simulation.28 The correlation between hydrogen bond persistence and binding free energy values supports the notion that hydrogen bonding significantly contributes to the strong binding affinities observed among high-scoring analogues. The ADMET profiling of all compounds provided valuable insights into their pharmacokinetic suitability for oral administration. The favorable water solubility (Log S) and high gastrointestinal absorption (GI%) values of TF8A, TF5A, TF1A, and TF7A indicated efficient intestinal uptake and systemic availability. Furthermore, the prediction of moderate blood–brain barrier (BBB) permeability suggested adequate central distribution potential without excessive neurotoxicity, a key advantage in designing safer Tafenoquine analogues.29 The majority of compounds exhibited low cytochrome P450 (CYP) inhibition potential, particularly toward CYP1A2, CYP2C19, CYP2C9, and CYP3A4, reducing the risk of metabolic interference and drug–drug interactions. However, partial inhibition of CYP2D6 observed in most derivatives, except TF1A and TF7A, warrants further metabolic characterization to assess interindividual variability in drug clearance. The non-hepatotoxic and non-sensitizing nature of several analogues (TF6A, TF7A, TF5A, and TF1A) reflects a favorable safety profile, crucial for preclinical evaluation. The predicted total clearance values indicated that these compounds are efficiently eliminated, minimizing the risk of bioaccumulation. Collectively, the combined docking, ADMET, and hydrogen bond occupancy results suggested that TF6A, TF5A, and TF8A exhibit strong binding affinity, favorable pharmacokinetic properties, and acceptable safety margins, making them promising candidates for further in vitro and in vivo antimalarial studies. These findings align with prior computational and pharmacological investigations highlighting the potential of 8-aminoquinoline-based scaffolds to inhibit key Plasmodium enzymes involved in parasite survival and replication. The ability of the designed Tafenoquine analogues to interact stably within the catalytic pocket of Plasmodium protease reinforces their potential as next-generation antimalarial agents.30
Hydrogen bond occupancy described the proportion of the molecular dynamics simulation during which a specific hydrogen bond remained intact, serving as an indicator of the strength and persistence of ligand–receptor interactions. Compounds exhibiting higher occupancy percentages demonstrated stronger and more stable hydrogen bonding with the active-site residues, thereby contributing to enhanced binding affinity and overall complex stability. Tafenoquine displayed moderate hydrogen bond occupancy with several key residues, whereas its analogues—particularly TF6, TF7, and TF8—showed higher and more sustained occupancy values throughout the simulation trajectory. These observations suggested that these derivatives formed stable hydrogen bond networks that persisted over time, providing structural reinforcement within the binding pocket. Conversely, analogues with lower occupancy values exhibited more transient hydrogen bond interactions, reflecting reduced conformational stability and weaker binding consistency. The hydrogen bond occupancy results strongly corroborated the molecular docking findings, confirming that TF6 and TF7 maintained robust and stable interactions with the Plasmodium protease receptor. The enhanced hydrogen bond persistence observed in these analogues likely contributed to their superior binding energies and improved pharmacodynamic potential compared to the parent compound, Tafenoquine.31 Further evaluation of drug-likeness using the Ghose, Veber, Egan, and Muegge rule sets revealed that all designed compounds satisfied acceptable physicochemical and pharmacokinetic criteria for oral drug candidates. Among them, TF1A and TF7A achieved the highest bioavailability scores, while TF8A and TF5A displayed slightly lower—but still favorable—values. Collectively, these data indicated that TF1A, TF7A, TF5A, and TF8A combined optimal binding affinity, favorable ADMET characteristics, and desirable bioavailability profiles, identifying them as promising lead candidates for further preclinical optimization and development as next-generation antimalarial agents.
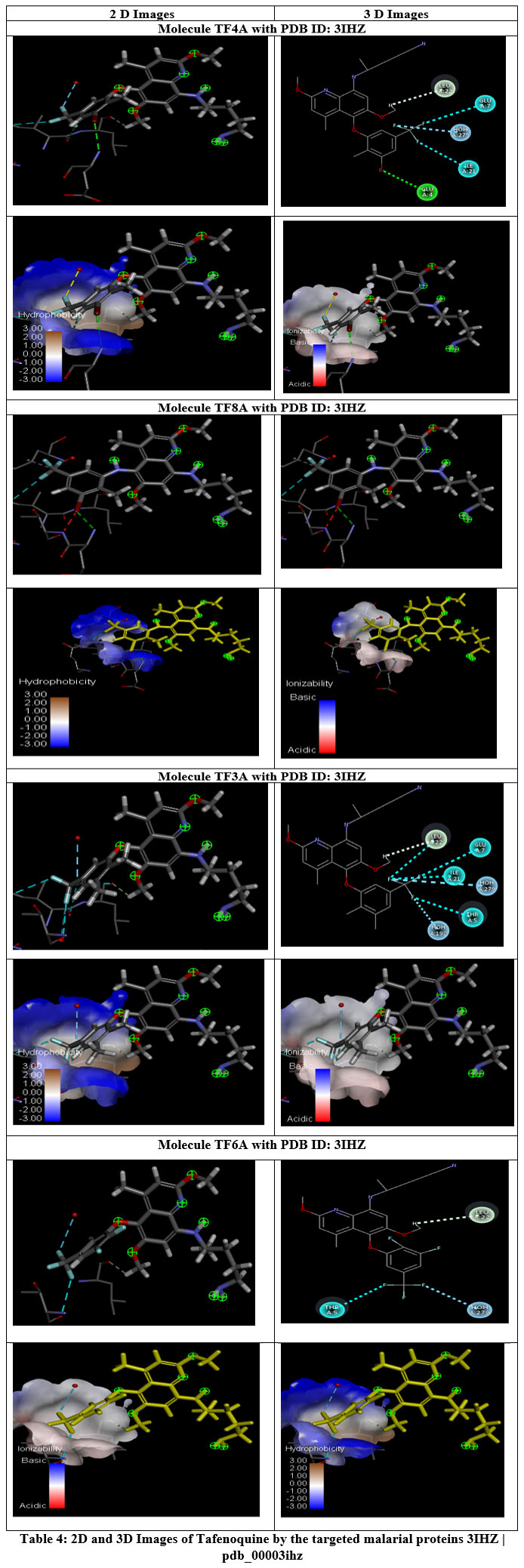 |
Table 4: 2D and 3D Images of Tafenoquine by the targeted malarial proteins 3IHZ | pdb_00003ihz
|
Table 5: Name of Amino acid, Hydrophobicity, pKa and Avg. Isotropic Displacement of Tafenoquine by the targeted malarial proteins 3IHZ | pdb_00003ihz proteins by Discovery Studio software’s.
| Name of Amino acid |
Hydrophobicity | pKa | Avg. Isotropic Displacement |
Molecular Docking |
| TF5A | -7.7 kcal/mol | |||
| GLU4 | -3.5 | 4.3 | 46.931 | |
| THR5 | -0.7 | 40.387 | ||
| LEU6 | 3.8 | 30.847 | ||
| GLU7 | -3.5 | 4.3 | 25.868 | |
| GLN8 | -3.5 | 22.918 | ||
| VAL9 | 4.2 | 15.341 | ||
| HIS10 | -3.2 | 6 | 15.858 | |
| LEU11 | 3.8 | 15.734 | ||
| THR12 | -0.7 | 18.181 | ||
| GLU13 | -3.5 | 4.3 | 24.961 | |
| ASP14 | -3.5 | 3.9 | 19.347 | |
| GLY15 | -0.4 | 17.21 | ||
| GLY16 | -0.4 | 14.205 | ||
| VAL17 | 4.2 | 12.873 | ||
| VAL18 | 4.2 | 12.447 | ||
| LYS19 | -3.9 | 10.4 | 14.089 | |
| THR20 | -0.7 | 15.337 | ||
| ILE21 | 4.5 | 19.655 | ||
| TF8A | -7.9 kcal/mol | |||
| GLY28 | -0.4 | 38.815 | ||
| GLU29 | -3.5 | 4.3 | 37.274 | |
| GLU30 | -3.5 | 4.3 | 37.339 | |
| ASN31 | -3.5 | 34.704 | ||
| ALA32 | 1.8 | 27.728 | ||
| PRO33 | -1.6 | 24.249 | ||
| LYS34 | -3.9 | 10.4 | 25.908 | |
| LYS35 | -3.9 | 10.4 | 25.61 | |
| GLY36 | -0.4 | 20.467 | ||
| ASN37 | -3.5 | 21.584 | ||
| GLU38 | -3.5 | 4.3 | 23.974 | |
| VAL39 | 4.2 | 17.1 | ||
| THR40 | -0.7 | 18.476 | ||
| VAL41 | 4.2 | 16.374 | ||
| HIS42 | -3.2 | 6 | 18.362 | |
| TYR43 | -1.3 | 10 | 14.868 | |
| VAL44 | 4.2 | 15.934 | ||
| GLY45 | -0.4 | 13.598 | ||
| LYS46 | -3.9 | 10.4 | 20.219 | |
| LEU47 | 3.8 | 15.96 | ||
| GLU48 | -3.5 | 4.3 | 17.833 | |
| SER49 | -0.8 | 19.09 | ||
| SER50 | -0.8 | 16.603 | ||
| GLY51 | -0.4 | 14.395 | ||
| TF6A | -8.5 kcal/mol | |||
| GLU4 | -3.5 | 4.3 | 46.931 | |
| THR5 | -0.7 | 40.387 | ||
| LEU6 | 3.8 | 30.847 | ||
| GLU7 | -3.5 | 4.3 | 25.868 | |
| GLN8 | -3.5 | 22.918 | ||
| VAL9 | 4.2 | 15.341 | ||
| HIS10 | -3.2 | 6 | 15.858 | |
| LEU11 | 3.8 | 15.734 | ||
| THR12 | -0.7 | 18.181 | ||
| GLU13 | -3.5 | 4.3 | 24.961 | |
| ASP14 | -3.5 | 3.9 | 19.347 | |
| GLY15 | -0.4 | 17.21 | ||
| GLY16 | -0.4 | 14.205 | ||
| VAL17 | 4.2 | 12.873 | ||
| VAL18 | 4.2 | 12.447 | ||
| LYS19 | -3.9 | 10.4 | 14.089 | |
| THR20 | -0.7 | 15.337 | ||
| TF1A | -8.2 kcal/mol | |||
| GLU4 | -3.5 | 4.3 | 46.931 | |
| THR5 | -0.7 | 40.387 | ||
| LEU6 | 3.8 | 30.847 | ||
| GLU7 | -3.5 | 4.3 | 25.868 | |
| GLN8 | -3.5 | 22.918 | ||
| VAL9 | 4.2 | 15.341 | ||
| HIS10 | -3.2 | 6 | 15.858 | |
| LEU11 | 3.8 | 15.734 | ||
| THR12 | -0.7 | 18.181 | ||
| GLU13 | -3.5 | 4.3 | 24.961 | |
| ASP14 | -3.5 | 3.9 | 19.347 | |
| GLY15 | -0.4 | 17.21 | ||
| GLY16 | -0.4 | 14.205 | ||
| VAL17 | 4.2 | 12.873 | ||
| VAL18 | 4.2 | 12.447 | ||
| LYS19 | -3.9 | 10.4 | 14.089 | |
| THR20 | -0.7 | 15.337 | ||
| ILE21 | 4.5 | 19.655 | ||
| LEU22 | 3.8 | 22.837 | ||
| ARG23 | -4.5 | 12 | 28.409 | |
| LYS24 | -3.9 | 10.4 | 32.306 | |
| GLY25 | -0.4 | 32.41 | ||
| GLU26 | -3.5 | 4.3 | 39.832 | |
| GLY27 | -0.4 | 39.082 | ||
| GLY28 | -0.4 | 38.815 | ||
| GLU29 | -3.5 | 4.3 | 37.274 | |
| GLU30 | -3.5 | 4.3 | 37.339 | |
| ASN31 | -3.5 | 34.704 | ||
| ALA32 | 1.8 | 27.728 | ||
Conclusion
The present investigation evaluated the antimalarial potential of Tafenoquine-derived analogues as selective inhibitors of the Plasmodium vivax protease (PDB ID: 3IHZ) using advanced computational techniques. Molecular docking analyses revealed that the derivatives TF1A, TF6A, TF8A, and TF5A exhibited stronger binding affinities toward the target protein than the parent molecule, Tafenoquine. The results of molecular dynamics simulations further verified that the respective ligand–receptor complexes maintained conformational stability and structural integrity throughout the simulation trajectory, demonstrating consistent interaction patterns within the active site. Comprehensive drug-likeness screening according to the criteria of Lipinski, Ghose, Veber, Egan, and Muegge indicated that several analogues, particularly TF2, TF6, and TF7, fulfilled the majority of physicochemical requirements and possessed favorable bioavailability scores. The ADMET analyses predicted low gastrointestinal absorption, non–blood–brain barrier permeability, and negligible cytochrome P450 enzyme inhibition, suggesting a low probability of adverse pharmacokinetic interactions and supporting their potential for safe oral administration. Furthermore, hydrogen bond occupancy evaluation, supported by Mcule 1-Click Docking validation, demonstrated that TF6 and TF8 formed persistent and stable hydrogen bonds with critical residues of the target protein, contributing to enhanced binding strength and complex stability compared with Tafenoquine. In summary, the integrated computational outcomes identified TF1, TF6, and TF8 as the most promising Tafenoquine analogues with superior binding affinities, optimal pharmacokinetic attributes, and desirable drug-likeness profiles. These compounds warrant further synthetic optimization and experimental validation through in vitro and in vivo approaches to confirm their antimalarial efficacy. Overall, this study provided valuable structural and pharmacological insights that may guide the rational design of next-generation antimalarial therapeutics, thereby supporting ongoing global malaria eradication initiatives.
Acknowledgement
The authors are thankful to Pravara Rural College of Pharmacy, Pravaranagar.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to Reproduce Material from Other Sources
Not Applicable
Author Contributions
Soniya Vaibhav Katore: In silico ADMET (Absorption, Distribution, Metabolism, Excretion)
Ajay Sanjay Mule: In silico ADMET (Absorption, Distribution, Metabolism, Excretion)
Neha Vilas Pagare: Drawing of Tafenoquine drug derivatives
Namrata Rajesh Adhav: SWISS ADMET Software Data
Harshali Narayan Anap: Docking images of 2D and 3D by Discovery Studio 2021
Rohit Jaysing Bhor: The active amino residues, bond length, bond category, bond type, ligand energies, 1-Click Docking software and Research drafting
References
- Fenta M, Kahaliw W. Evaluation of antimalarial activity of hydromethanolic crude extract and solvent fractions of the leaves of Nuxia congesta R. Br. Ex Fresen (Buddlejaceae) in Plasmodium berghei infected mice. J Exp Pharmacol. 2019;11:121–134. doi: 10.2147/JEP.S230636.
CrossRef - Geleta G, Ketema T. Severe malaria associated with Plasmodium falciparum and P. vivax among children in Pawe hospital, Northwest Ethiopia. Malar Res Treat. 2016;2016:1240962. doi: 10.1155/2016/1240962.
CrossRef - Conrad MD, Rosenthal PJ. Antimalarial drug resistance in Africa: the calm before the storm? Lancet Infect Dis. 2019;19(10):e338–ee51. doi: 10.1016/S1473-3099(19)30261-0.
CrossRef - Oboh MA, Ndiaye D, Antony HA, Badiane AS, Singh US, Ali NA, et al. Status of artemisinin resistance in malaria parasite Plasmodium falciparum from molecular analyses of the Kelch13 gene in southwestern Nigeria. Biomed Res Int. 2018;2018:2305062. doi: 10.1155/2018/2305062.
CrossRef - Antimalarial drug resistance: a threat to malaria elimination. Cold Spring Harb Perspect Med. 2017;7(7):a025619. doi: 10.1101/cshperspect.a025619.
CrossRef - Uzor PF. Alkaloids from plants with antimalarial activity: a review of recent studies. Evid Based Complement Alternat Med. 2020;2020:8749083. doi: 10.1155/2020/8749083.
CrossRef - Singh R, Bhardwaj V, Purohit R. Identification of a novel binding mechanism of quinoline based molecules with lactate dehydrogenase of Plasmodium falciparum. J Biomol Struct Dyn. 2020;39:1–9. doi: 10.1080/07391102.2020.1711809.
CrossRef - Ntie-Kang F, Onguéné PA, Lifongo LL, Ndom JC, Sippl W, Mbaze LM. The potential of anti-malarial compounds derived from African medicinal plants, part II: a pharmacological evaluation of non-alkaloids and non-terpenoids. Malar J. 2014;13:81. doi: 10.1186/1475-2875-13-81.
CrossRef - Jones RA, Panda SS, Hall CD. Quinine conjugates and quinine analogues as potential antimalarial agents. Eur J Med Chem. 2015;97:335–355. doi: 10.1016/j.ejmech.2015.02.002.
CrossRef - Brown GD. The biosynthesis of artemisinin (Qinghaosu) and the phytochemistry of Artemisia annua L. (Qinghao) Molecules. 2010;15(11):7603–7698. doi: 10.3390/molecules15117603.
CrossRef - Ghosh S, Ahire M, Patil S, Jabgunde A, Bhat Dusane M, Joshi BN, et al. Antidiabetic activity of Gnidia glauca and Dioscorea bulbifera: potent amylase and glucosidase inhibitors. Evid Based Complement Altern Med. 2012;2012:929051. doi: 10.1155/2012/929051.
CrossRef - Mbiantcha M, Kamanyi A, Teponno R, Tapondjou A, Watcho P, Nguelefack TB. Analgesic and anti-inflammatory properties of extracts from the bulbils of Dioscorea bulbifera L. var sativa (Dioscoreaceae) in mice and rats. Evid Based Complement Altern Med. 2011;2011:912935. doi: 10.1155/2011/912935.
CrossRef - Savi A, Calegari G, Santos V, Pereira E, Teixeira S. Chemical characterization and antioxidant of polysaccharide extracted from Dioscorea bulbifera. J King Saud Univ Sci. 2018;32:636–642. doi: 10.1016/j.jksus.2018.09.002.
CrossRef - Chaniad P, Wattanapiromsakul C, Pianwanit S, Tewtrakul S. Anti-HIV-1 integrase compounds from Dioscorea bulbifera and molecular docking study. Pharm Biol. 2016;54(6):1077–1085. doi: 10.3109/13880209.2015.1103272.
CrossRef - Phuwajaroanpong A, Chaniad P, Horata N, Muangchanburee S, Kaewdana K, Punsawad C. In vitro and in vivo antimalarial activities and toxicological assessment of Pogostemon cablin (Blanco) Benth. J Evid Based Integr Med. 2020;25:2515690×20978387. doi: 10.1177/2515690X20978387.
CrossRef - Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother. 1979;16(6):710–718. doi: 10.1128/AAC.16.6.710.
CrossRef - Tahghighi A, Mohamadi-Zarch S-M, Rahimi H, Marashiyan M, Maleki-Ravasan N, Eslamifar A. In silico and in vivo anti-malarial investigation on 1-(heteroaryl)-2-((5-nitroheteroaryl)methylene) hydrazine derivatives. Malaria J. 2020;19(1):231. doi: 10.1186/s12936-020-03269-7.
CrossRef - Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015;43(W1):W443–W4W7. doi: 10.1093/nar/gkv315.
CrossRef - Fairhurst RM, Dondorp AM. Artemisinin-resistant Plasmodium falciparum malaria. Microbiol Spectr. 2016;4(3):409–429. doi: 10.1128/microbiolspec.EI10-0013-2016.
CrossRef - Pattaradilokrat S, Sawaswong V, Simpalipan P, Kaewthamasorn M, Siripoon N, Harnyuttanakorn P. Genetic diversity of the merozoite surface protein-3 gene in Plasmodium falciparum populations in Thailand. Malaria J. 2016;15(1):517. doi: 10.1186/s12936-016-1566-1.
CrossRef - Ganesh D, Fuehrer HP, Starzengrüber P, Swoboda P, Khan WA, Reismann JA, et al. Antiplasmodial activity of flavonol quercetin and its analogues in Plasmodium falciparum: evidence from clinical isolates in Bangladesh and standardized parasite clones. Parasitol Res. 2012;110(6):2289–2295. doi: 10.1007/s00436-011-2763-z.
CrossRef - Li Y, Yao J, Han C, Yang J, Chaudhry MT, Wang S, et al. Quercetin, inflammation and immunity. Nutrients. 2016;8(3):167. doi: 10.3390/nu8030167.
CrossRef - Zahoor M, Shafiq S, Ullah H, Sadiq A, Ullah F. Isolation of quercetin and mandelic acid from Aesculus indica fruit and their biological activities. BMC Biochem. 2018;19(1):5. doi: 10.1186/s12858-018-0095-7.
CrossRef - Lee HN, Shin SA, Choo GS, Kim HJ, Park YS, Kim BS, Kim SK, Cho SD, Nam JS, Choi CS, Che JH, Park BK, Jung JY. Anti-inflammatory effect of quercetin and galangin in LPS-stimulated RAW264.7 macrophages and DNCB-induced atopic dermatitis animal models. Int J Mol Med. 2018;41(2):888–898. doi: 10.3892/ijmm.2017.3296.
CrossRef - Xu D, Hu MJ, Wang YQ, Cui YL. Antioxidant activities of quercetin and its complexes for medicinal application. Molecules. 2019;24(6):1123. doi: 10.3390/molecules24061123.
CrossRef - White NJ. “The Antimalarial Activity of Tafenoquine in Falciparum Malaria.” Clinical Infectious Diseases. 2023;76(11):1928-1929. doi:10.1093/cid/ciad079.
CrossRef - Watson J, et al. “The Clinical Pharmacology of Tafenoquine in the Radical Cure of Plasmodium vivax Malaria: an Individual Patient Data Meta-analysis.” 2023;12:e83433. doi:10.7554/eLife.83433.
CrossRef - Syahri J, Hilma R, Ali AH, et al. “Chalcone Mannich base derivatives: synthesis, antimalarial activities against Plasmodium knowlesi, and molecular docking analysis.” RSC Advances. 2023;13:36035-36047. doi:10.1039/D3RA05361J.
CrossRef - Chen Y, Yang W-H, Chen H-F, Huang L-M, Gao J-Y, Lin C-W, et al. Tafenoquine and its derivatives as inhibitors for the severe acute respiratory syndrome coronavirus 2. J Biol Chem. 2022 Mar;298(3):101658. doi:10.1016/j.jbc.2022.101658.
CrossRef - She P, Yang Y, Li L, Li Y, Liu S, Li Z, Zhou L, Wu Y. Repurposing of the antimalarial agent tafenoquine to combat MRSA. mSystems. 2023 Dec;8(6):e0102623. doi:10.1128/msystems.01026-23.
CrossRef - Manen-Freixa L, Moliner-Cubel S, Gamo F-J, Crespo B, Borrell JI, Teixidó J, Estrada-Tejedor R. Exploring the unexplored chemical space: rational identification of new tafenoquine analogs with antimalarial properties. Bioorg Chem. 2024;148:107472. doi:10.1016/j.bioorg.2024.107472.
CrossRef - Fenta, M., and W. Kahaliw. “Evaluation of Antimalarial Activity of Hydromethanolic Crude Extract and Solvent Fractions of the Leaves of Nuxia congesta Br. Ex Fresen (Buddlejaceae) in Plasmodium berghei-Infected Mice.” Journal of Experimental Pharmacology, vol. 11, 2019, pp. 121–134. https://doi.org/10.2147/JEP.S230636
CrossRef
Abbreviations List
GLU: Glutamic acid; THR: Threonine; LEU: Leucine; GLN: Glutamine; VAL: Valine; HIS: Histidine; ASP: Aspartic acid GLY: Glycine; LYS: Lysine; ILE: Isoleucine; ASN: Asparagine; ALA: Alanine; PRO: Proline; SER: Serine; TYR; Tyrosine; GI absorption Gastrointestinal absorption; BBB: Blood–brain barrier; Pgp: P-glycoprotein; CYP: Cytochrome P450 enzyme; CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4: Specific isoforms of Cytochrome P450 enzymes involved in drug metabolism; log Kp: Skin permeation coefficient (cm/s); Lipinski violations: Number of violations of Lipinski’s Rule of Five (drug-likeness rule); ADMET: Absorption, Distribution, Metabolism, Excretion, and Toxicity; TPSA: Topological polar surface area; Log P: Octanol–water partition coefficient; SMILES: Simplified Molecular Input Line Entry System; 3IHZ: PDB ID of Plasmodium vivax protease used as the target protein
Accepted on: 06-01-2026
Second Review by: Dr. Sunil Chaudhryand Dr. Saeed Kewedar
Final Approval by: Dr. Wagih Ghannam








![]()
![]()
