

Manuscript accepted on : 20 June 2018
Published online on: 29-06-2018
Plagiarism Check: Yes
Akanksha Joshi , Rajesh Kumar and Archit Sharma
, Rajesh Kumar and Archit Sharma
Department of Biotechnology, University Institute of Engineering and Technology, Kurukshetra University Kurukshetra, India.
Corresponding Author E-mail: archit.sharma786@gmail.com
DOI : http://dx.doi.org/10.13005/bbra/2650

ABSTRACT: Glycogen synthase kinase 3 β (GSK-3 Beta) is a potential target for developing an effective therapeutic effect in Alzheimer's disease (AD). Currently, no such drug or molecules has been found till date which can cure AD completely. Few drugs such as acetylcholinesterase inhibitors and memantine are ineffective in the later stages of the disease. Therefore, with the advancements in computational biology approaches, it is possible to combat alzheimer’s disease by targeting one of the kinases i.e. GSK-3 β involved in hyper phosphorylation of tau (a reliable marker of neurodegenerative disorders). In this study, we have carried out alzheimer’s structure-based drug designing with GSK-3 β. By applying appropriate docking methodology, we have identified few plant-derived compounds which show enhanced target selectivity than the conventional alzheimer's drug (such as memantine). Here we enumerate the comparison among the current and future AD therapy on the basis of their binding affinities. As a result, a large library of compounds has been screened as potent drug targets. It was also observed that withanolide–A (extracted from roots of withania somnifera) has the potential to emerge as the eventual drug for the AD. Moreover, few other phytocompounds such as celastrol, kenpaullone, quercetin, alsterpaullone have also shown enhanced activity in the decreasing order of their binding affinities.
KEYWORDS: Alzheimer’s disease (AD); Drug Designing; Glycogen Synthase Kinase 3 β (GSK-3 β); Hyperphosphorylation of tau, Memantine; Withanolide-A (Ashwagandha)
Download this article as:| Copy the following to cite this article: Joshi A, Kumar R, Sharma A. Molecular Docking Studies, Bioactivity Score Prediction, Drug Likeness Analysis of GSK-3 Β Inhibitors: A Target Protein Involved in Alzheimer’s Disease. Biosci Biotech Res Asia 2018;15(2). |
| Copy the following to cite this URL: Joshi A, Kumar R, Sharma A. Molecular Docking Studies, Bioactivity Score Prediction, Drug Likeness Analysis of GSK-3 Β Inhibitors: A Target Protein Involved in Alzheimer’s Disease. Biosci Biotech Res Asia 2018;15(2). Available from: https://www.biotech-asia.org/?p=30221 |
Introduction
Alzheimer’s is a form of dementia associated with loss of memory or cognition, serious enough to interfere with the daily life. According to the report prepared by Alzheimer’s and related disorders society of India in 2010, there are 3.7 million Indians suffering with dementia while the numbers are anticipated to bifold by 2030. The present Alzheimer’s therapies are ineffective to treat symptoms such as cognition especially in the later stages( www.alz.org). For more than a decade, researchers have found ‘tau’ protein as one of the causes other than the Beta-amyloid plaques (Underwood 2016). Due to the lack of the utility of amyloid-β-aspired approach in Phase 111 clinical trials, it was prerequisite to consider alternative drug discovery strategies for alzheimer’s (Folch et al. 2016). Solanezumab, a drug which acts on amyloid β protein failed some pivotal clinical trials. A number of normal patients have been found with amyloid deposists in their brain. It was anticipated that amyloid beta deposition is an anomaly of aging and does not correlate with the AD progression(Kametani et al. 2018). Therefore a study adopted policies directed at reducing misfolded tau (due to hyperphosphorylation) which is one of the disease-causing agents (Bruden et al. 2009). Tau is liable to be the more superior target than the amyloid β as it coordinates efficiently with cognitive impairement, provided clinical symptoms are tangible (Congdon et al. 2018). Hence, we can affirm that tau is a reliable marker of the neurodegenerative process.Changes in tau confirmation could lead to excessive phosphorylation resulting in the formation of neurotoxic aggregates and tau-mediated neurodegeneration (Dixit et al. 2008). Excessive phosphorylation of tau leads to the organization of paired helical fragments (PHF’s) due to the loss of affinity with microtubules and they bind with one another which further aggregates in neurofibrillary tangles via. post-translational modifications (Avila 2006; Martin et al. 2011,2013). Thus, there is a strong correlation between abnormal phosphorylation and self-aggregation of tau (Guo et al. 2017).One of the studies demonstrated that dephosphorylation of the hyperphosphorylated tau converts abnormal tau protein into a normal like protein which then regulates microtubule assembly( Iqbal et al. 2010).Therefore abrogating the abnormal tau and recovery of the microtubule organization are the most promising therapeutic interventions to combat AD (Figure 1).
 |
Figure 1: A. Factors and Major Causes for Alzheimer’s Disease; B. Events that are involved in tau mediated neurodegeneration
|
Normal tau is hyperphosphorylated by; 1) Over activation of kinases such as GSK-3β, 2) inactivation of phosphatases such as PPA, PP2B
Tau hyperphosphorylation and accumulation of insoluble aggregates results in formation of paired helical fragments (PHF’s) followed by Neurofibrillary tangles(NFT’s).
Formation of neurotoxic aggregates is the major hallmark in tau-mediated neurodegeneration
Such a pathological event ultimately leads to Alzheimer’s.
Recovery of abnormal tau into a normal like protein by using GSK-3β as a drug target- Inhibiting GSK-3β with the aid of novel therapeutic phytocompounds is the most promising therapeutic intervention.
Targeting tau mediated neurodegeneration is one such approach to combat AD and provide neuroprotection.
Protein kinases such as Glycogen synthase kinase 3 β have been identified as promising drug targets because of their involvement in breakthrough of AD pathways like pathophysiological tau protein phosphorylation or tau hyperphosphorylation. The sound interdependence of tau phosphorylation and pathology has led to the search for Tau protein kinase inhibitor such as GSK3- β which phosphorylates tau and also plays a causative role in amyloid pathway. Hereafter, acting as a potential therapeutic agent (Medina, 2018). Kinases are involved in tau phosphorylation and phosphatases reverse this action.GSK-3 is encoded by two genes: GSK-3 α, positioned on chromosome 19 and GSK-3 β, located on chromosome 2. GSK-3 is ubiquitously expressed in mammals as well as in yeast (Medina et al. 2011).GSK-3 is a serine/threonine protein kinase.GSK-3 β has a molecular mass of 46-47 kDa existing of 433 and 420 amino acids in humans and mouse respectively. The protein contains an N- terminal domain, a kinase domain, and a C-terminal domain. The substrate Binding domain (BD) provides GSK-3 β specific binding sites for the tumor supressor p53 and other protein complexes (Atlas of Genetics and Cytogenetics in Oncology and Haematology). It has been reported that 31% of the pathological phosphorylation sites of tau protein are phosphorylated by GSK-3 β (Martin et al. 2013). Moreover, GSK-3 β results in the neuronal decline in the AD because of the fact that it is a causal mediator of apoptosis. Increased level of such protein eventuated in the autopsy evaluation of brain of alzheimer’s victims (Pei et al. 1997). According to the ‘GSK-3 hypothesis of AD’, tau hyperphosphorylation, memory impairment and enhanced β- amyloid production is due to the overexpression of GSK-3, all of which are characteristic features of the disease. If this hypothesis is consolidated then inhibition of GSK-3 β by novel inhibitors provides a better pathway against the effect of this destructing disorder (Hooper et al. 2008).
There are no such drugs/ treatments available that can cure AD completely. However, there are several medications developed that can temporarily attenuate the symptoms. The U.S Food and Drug Administration (FDA) have affirmed two medications- acetylcholinesterase inhibitors and Memantine. Drugs such as Tacrine, rivastigmine, galantamine, and donepezil are widely used conventional drugs to treat AD (Islam et al. 2013). Therefore, traditional drugs such as memantine and donepezil are being extendedly used as a reference in molecular docking studies. The antioxidant ability of flavonoids (obtained from plants) has encouraged their use as an agent to improve neurological health. Increasing evidence shows their ability to improve brain function such as memory and learning by interacting with cellular as well as molecular components of the brain resulting in enhanced neuronal function and induce neurogenesis (Spencer 2010; Baptista et al. 2014). Hence, the objective of eventual AD therapy is to develop such possible compounds that could abrogate the tau protein and thereby it can be utilized for the remedy of neurodegenerative diseases (Schneide et al. 2008). The study related to the AD is focused more towards the traditional medicinal plants and its components such as Withania somnifera. It is commonly called Ashwagandha, Indian ginseng and wind cherry have been an important herb in Indigenous and ayurvedic medical system. Historically, the plant has been used therapeutically for boosting the brain function including memory retrieval. Thus has a cognition promoting effect in adults and children (Singh et al. 2011). It consists of two components: withanolides, withanamides. Withanolide A is extracted from the roots of the plant and promotes antioxidant properties that protect nerve cells from harmful free radicals. Withanolides have also been used for the treatment of AD (Khan et al. 2016). Instead of the root extract, a study also suggested fruits and leaves of Egyptian plant have strong antioxidant activity (Mahrous et al. 2017). Drug research is of utmost importance in the field of medicine. Consequently, the manipulation of computers to predict the efficiency of binding of a set of molecules or ligands with the target is an important element of drug development process. Autodock 4.2 which is a molecular modeling simulation software is widely used for virtual screening (Collignon et al. 2011). In this, the ligand being docked was kept as flexible while target protein was kept as rigid. Autodock Tools was used to prepare, run the protein and ligand files separately as well as analyzes the docking simulations.
Material and Methods
An array of tools and software such as AutoDock Tools, Pymol, Ligplot+ are required to analyze the receptor GSK-3β and review the binding energies of various protein-ligand complexes. Glycogen synthase kinase 3 Beta (GSK-3β) enzyme with PDB code 1J1C sequence was obtained from protein data bank (www.pdb.org/pdb/). To get vision of the intermolecular interactions, the molecular docking studies were done for the below-mentioned phytoconstituents and taking memantine (3, 5-dimethyladamantan-1-amine) as a reference which is a FDA approved drug. PyMOL which is an OpenGL based molecular visualization system is used to visualize 3D structure of our protein GSK-3β. The protein is represented as a cartoon and water molecules were removed from it along with the hydrogens.The sequence can be displayed from the dialog box and heteroatoms as well as already present ligand were also removed (as shown in figure 2). An extensive literature survey was done to find the possible inhibitors of GSK-3 β. Various 3D structures of the ligands were drawn from the PubChem (https://pubchem.ncbi.nlm.nih.gov/ ).Using SMILES translator which is an online tool for generating files from mol format to PDB format (i.e. into the 3D models of the molecules).Binding sites were determined from the PDB(www.pdb.org/pdb/) by introspecting the binding pockets of the already present ligand i.e. ADP. ADP forms a complex with GSK-3β via flexible residues which are as follows: Val 70A, Lys 85A, Ala 83A, Asp133A, Val 135A, Gln185A.Water molecules were removed, Gasteiger charges were added, nonpolar hydrogens were merged to prepare the protein file for docking stimulation. Information is added to the ligand PDB file such as the rigid roots of each ligand were defined, torsions were chosen and rotatable bonds were selected. Than ligand file was saved as pdbqt. Rather than treating the protein entirely as the rigid shape, a part of it was made flexible and then flexible residues were added. They were as follows: Val 70A, Lys 85A, Ala 83A, Asp133A, Val 135A, and Gln185A (Figure 4). Now rotatable bonds were selected in each flexible residue separately. The flexible residues were saved; Flexible PDBQT as GSK-3_ flex and the rigid PDBQT as GSK-3_rigid. The protein file was further prepared by selection of grid parameters, map types and it was checked that all atoms in the ligand were represented. Finally, the grid box within which we are going to search was selected and size of the box, its location, and the number and spacing of the points within it that will be tested (as shown in figure 3). The grid box was centralized on the catalytic site of the GSK3-B which was determined by inspecting the PDB file and the output was saved. Now by further selecting the docking parameters as default, the docking parameter file was prepared. Lamarckian Genetic algorithm (GA) was selected and the final output was saved as dlg (docking ligand file). Autodock was launched from the autodock tools.
Binding energy of the individual protein-ligand complex was pre-calculated and obtained in a dlg file using the following formula, Binding energy = P+Q+R-S (Shown in table 1) where, P = final intermolecular energy + van der Walls energy (vdW) + hydrogen bonds + desolvation energy + electrostatic energy (kcal/mol), Q = final total internal energy (kcal/mol), R = torsional free energy (kcal/mol), S = unbound system’s energy (kcal/mol) (Madeswaran et al. 2013).
Finally, by using Ligplot +, 2-D representation of protein-ligand complex was generated which was obtained as a PDB file from Autodock Tools.
Bioactivity Score Prediction
Smiles notation of the selected compounds were fed into the Molinspiration Virtual Screening online software (www.molinspiration.com) and two separate dialoug boxes will appear on the screen representing the bioactivity score(GPCR ligands, kinase inhibitors, ion channel modulators, enzymes and nuclear receptors)and the physiochemical properties of the ligands using Lipinski’s rule(Log P, Total polar surface area, number of hydrogen bond donors and acceptors, molecular weight, number of atoms, number of rotatable bonds etc.).The bioactivity score and druglikeness properties of the selected ligands were compared against the conventional drug memantine.
Evaluation of Physiochemical Properties (“drug likeness”) using Lipinski’s Rule of Five
Lipinski’s rule of five also known as rule of five ( RO5) is used to evaluate whether a compound with certain physiochemical activity has physical and chemical properties that would make it orally active , i.e. it helps in the estimation of parameters such as; A – absorption , D-Distribution, M-Metabolism and E- Excretion (Khan et al. 2017).
Results and Discussions
To identify the efficiency of molecular binding between the ligand and the receptor, molecular docking analysis was accomplished using Autodock 4. All the ligands were retrieved from Pub Chem whereas the protein GSK-3 Beta was fetched from PDB (i.e. Protein Data Bank). Two main programs involved in AutoDock Tools are Autodock for docking of the ligand within the set of grids(i.e. within the binding site) in the target protein and Autogrid for selection of grid parameters, size of the box, its location etc (http://autodock.scripps.edu/). Autodock 4.2 is the ultimate current version which is based upon the Lamarckian genetic algorithm, a hybrid algorithm comprising of both the genetic as well as local search unlike genetic algorithm which performs global search and is more enhanced. (Madeswaran et al. 2011). It is more accurate than previous version AD3.0. Unlike AD3.0, Autodock 4.2.6 ( henceforth AD4.2) and AutoDock Vina 1.1.2 (henceforth AD Vina) have upgraded results and improved elucidation (Alvarez et al. 2017). This methodology is especially adequate for protein-ligand docking in which we anticipate the locus and orientation of a small molecule when it is bound to a protein receptor.It is used to select likely drug candidates. Typically, ligand is a drug candidate (ion / molecule) that binds to a macromolecule which is the protein or receptor of the known three-dimensional structure. In this docking simulation, the ligands being docked were kept as flexible while target protein was kept as rigid and thereby the ligands were imbeded into the catalytic domain of the protein. The ligand is allowed to bind into groove of the receptor in various possible conformations. The resulted free energy of binding was compared with the FDA approved drug Memantine. The docking information is obtained in various parameters such as: hydrogen bond ( delta G H-bond ), electrostatic (delta G elec), torsional free energy(delta G torsional) dispersion and repulsion(delta G vdw), desolvation (delta G dissolv), total internal energy(delta G total) and unbound system’s energy ( delta G unb). It was interpreted from the docking results that the more negative the binding affinity/ energy is, more stable the complex is formed. The binding energy= energy of complex- the energy of ligand- the energy of receptor. This formula depicts that a complex has lower potential energy than its constituent parts. Hydrogen bonds play a critical role as they regulate protein-ligand specificity (Motiejunas et al. 2007). The main objective of this approach is to find the complex of high stability and optimized conformation As shown in figure 5, among all the ligands only one compound i.e. Quercetin has 6 Hydrogen bonds (H-bonds) while 4 hydrogen bonds were formed in Daidzein, Withanolide-A and Epicatechin-5-gallate whereas 3 were found in Kaempferol, Convoline, Pelargonidin, Luteolin. Compounds like Memantine, 3f8, Curcumin, Morin, Resveratrol have 2 H-bonds. There were some compounds with one hydrogen bond namely: Alsterpaullone, Celastrol, Celapanin, Kenpaullone, Paniculatine, Nicotine, apigenin, Scopoletin, Donepezil, Glycitein. Few others like Melatonin, Betaine,Catechin+, Tideglusib, Tdzd-8 have 0 H-bonds. Table 1 shows the highest docking score of Withanolide-A (-5.37) and has 4-H bonds and memantine has docking score of -3.57(H bonds=2). About 6 ligands have shown better score than the memantine. The docking scores of all the ligands are listed in table 1. The residues involved in formation of H-bonds are Asp700, Asp681, Asp264, Asp260, Asn686, Asn564, Cys699, Cys718, Ser703, Lys683 (Table 2). The bioactivity score parameters as well as drug likeness
Properties are listed in table 3 and table 4 respectively. Drug likeness is a concept used in drug design to determine for how” drug like” a particular molecule is with respect to the known drug. It is estimated using the molecular structure even before it has been synthesized and tested. It has been known that a compound has poor absorption if 1. It has more than 5 hydrogen bond donors (determines the solubility of compound with water) 2. It has more than 10 hydrogen bond acceptors 3. Molecular weight is less than 500(smaller molecules have better diffusion ability) 4. logP is less than 4.15( used to determine the solubility of the potent oral drug). None of our compounds violates this basis of drug likeness. Lipinski’s rule of five states that, an orally active drug has no more than one violation of the above mentioned criteria. If the bioactive score is >0 than the compound is active, -5.0-0.00 is moderate active, and <-5.0 is inactive. All our compounds have shown good bioactive score (Khan T et al. 2017).
Table 1: List showing docking scores of phytocompounds
| Name of ligands | Final intermolecular energy (kcal/mol) | Final total internal energy (kcal/mol) | Torsional free energy (kcal/mol) | Unbound system’s energy (kcal/mol) | The estimated free energy of binding kcal/mol) | Estimated imbibition constant; Ki | No of hydrogen bonds |
| Memantine | -3.87 | +2.04e+07 | +0.30 | +2.04e+07 | -3.57 | 2.40 mM | 2 |
| Celastrol | -5.62 | +2.04e+07 | +0.89 | +2.04e+07 | -4.73 | 0.342 mM | 1 |
| Kennpaulone | -4.49 | +2.04e+07 | +0.00 | +2.04e+07 | -4.49 | 0.51mM | 1 |
| 3f8 | -3.87 | +2.04e+07 | +0.89 | +2.04e+07 | -2.98 | 6.57mM | 2 |
| Curcumin | -5.49 | +2.04e+07 | +2.98 | +2.04e+07 | -2.51 | 14.48mM | 2 |
| Paniculatine | -5.63 | +2.04e+07 | +2.39 | +2.04e+07 | -3.24 | 4.20mM | 1 |
| Quercitine | -6.16 | +2.04e+07 | +1.79 | +2.04e+07 | -4.37 | 0.62mM | 6 |
| Nicotine | -2.41 | +2.04e+07 | +0.30 | +2.04e+07 | -2.11 | 28.27mM | 1 |
| Apigenin | -4.31 | +2.04e+07 | +1.19 | +2.04e+07 | -3.12 | 5.17mM | 1 |
| Scopoletin | -2.22 | +2.04e+07 | +0.60 | +2.04e+07 | -2.22 | 23.70mM | 1 |
| Kaempferol | -4.80 | +2.04e+07 | +1.49 | +2.04e+07 | -3.31 | 3.74mM | 3 |
| Convoline | -5.09 | +2.04e+07 | +1.79 | +2.04e+07 | -3.30 | 3.84mM | 3 |
| Withanolide A | -6.56 | +2.04e+07 | +1.19 | +2.04e+07 | -5.37 | 0.11mM | 4 |
| Melatonin | -3.22 | +2.04e+07 | +1.19 | +2.04e+07 | -2.03 | 32.75mM | 0 |
| Betaine | -2.22 | +2.04e+07 | +0.60 | +2.04e+07 | -1.63 | 64.35mM | 0 |
| Morin | -4.98 | +2.04e+07 | +1.79 | +2.04e+07 | -3.19 | 4.62mM | 2 |
| Alsterpaullone | -4.48 | +2.04e+07 | +0.30 | +2.04e+07 | -4.18 | 0.86mM | 1 |
| Tideglusib | -4.48 | +2.04e+07 | +0.89 | +2.04e+07 | -3.59 | 2.34mM | 0 |
| Tdzd-8 | -3.27 | +2.04e+07 | +0.60 | +2.04e+07 | -2.67 | 11.06mM | 0 |
| Donepezil | -4.12 | +2.04e+07 | +1.79 | +2.04e+07 | -2.33 | 19.52mM | 1 |
| Celapanin | -4.54 | +2.04e+07 | +2.98 | +2.04e+07 | -1.56 | 71.67mM | 1 |
| Glycitein | -3.37 | +2.04e+07 | +1.19 | +2.04e+07 | -2.18 | 25.33mM | `1 |
| Luteolin | -4.74 | +2.04e+07 | +1.49 | +2.04e+07 | -3.25 | 4.17mM | 3 |
| Pelargonidin | -4.73 | +2.04e+07 | +1.49 | +2.04e+07 | -3.24 | 4.23mM | 3 |
| Catechin (+) | -4.59 | +2.04e+07 | +1.79 | +2.04e+07 | -2.80 | 8.86 mM | 0 |
| Myrecetin | -4.81 | +2.04e+07 | +2.09 | +2.04e+07 | -2.72 | 10.12mM | 5 |
| Epicatechin-5-gallate | -5.54 | +2.04e+07 | +3.28 | +2.04e+07 | -2.26 | 21.98mM | 4 |
| Daidzein | -5.22 | +2.04e+07 | +2.68 | +2.04e+07 | -2.53 | 13.90mM | 4 |
| Resveratrol | -4.70 | +2.04e+07 | +1.49 | +2.04e+07 | -3.21 | 4.46mM | 2 |
 |
Figure 2: Overall domain structure of GSK-3β
|
A. Cartoon diagram of GSK-3β (prepared with pymol) showing: Protein Kinase domain (blue), active residues (pink), and the protein chain ( olive green)
B. Domain organization of GSK-3β
 |
Figure 3: Preparation of protein file (by setting grid parameters)
|
Spacing = 0.375
X center = 30.852
Y center =-6.308
Z center = -30.219
A. The box represents the grid box which is centered on the active site residues present on the protein
B. The spheres inside the box represent the active site residues.
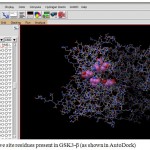 |
Figure 4: Active site residues present in GSK3-β (as shown in Auto Dock)
|
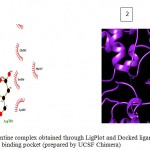 |
Figure 5a
|
Olive green dashes depict hydrogen bonds formed between receptor and drug molecule along with their distance in Angstrom.
Blue line depicts hydrogen bonds. Some important residues in the vicinity of ligand are Asp264.A, Asp700.B, Asp681.B, Lys 683.B, Asn686.B, and Ser147.A.
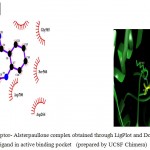 |
Figure 5b
|
Olive green dashes depict hydrogen bonds formed between receptor and drug moleculealong with their distance in Angstrom.
Blue line depicts hydrogen bonds .Some important residues in the vicinity of ligand are Lys585.B, Asp700.B, Ser566.B, Asp264.A
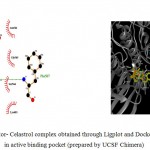 |
Figure 5c
|
Olive green dashes depict hydrogen bonds formed between receptor and drug moleculealong with their distance in Angstrom.
Blue line depicts hydrogen bonds .Some important residues in the vicinity of ligand are Gln685.B, Lys683.B, Asp260.A , Asn 564.B, Asp 700.B, Ser 566.
 |
Figure 5d
|
Olive green dashes depict hydrogen bonds formed between receptor and drug molecule along with their distance in Angstrom.
Blue line depicts hydrogen bonds.Some important residues in the vicinity of ligand are Gln730.B, Ser703.B, Asp681.B, Lys683.B, Gln685.B, Asp260.A, Val263.A, and Tyr716.B.
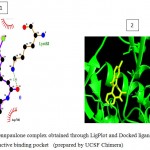 |
Figure 5e
|
Olive green dashes depict hydrogen bonds formed between receptor and drug molecule along with their distance in Angstrom.
Blue line depicts hydrogen bonds .Some important residues in the vicinity of ligand are Thr638.B, Gln685.B, Lys683.B, and Lys585.B.
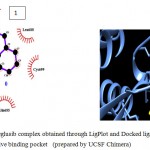 |
Figure 5f
|
Olive green dashes depict hydrogen bonds formed between receptor and drug molecule along with their distance in Angstrom.
Blue line depicts hydrogen bonds .Some important residues in the vicinity of ligand are Cys699.B, Gln 685.B, Asn686.B, Lys683.B, and Asp264.A.
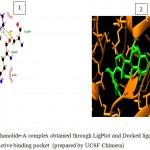 |
Figure 5g
|
Olive green dashes depict hydrogen bonds formed between receptor and drug molecule along with their distance in Angstrom.
Blue line depicts hydrogen bonds. Some important residues in the vicinity of ligand are Asp700.B, Cys699.B, Gln685.B, Asp260.A, Ser261.A, Asp264.A, Ser566.B.Asn 686.B, Cys683.B, and Lys585.B.
Figure 5: 2D representation of protein-ligand complex and docking interaction of GSK-3β with the ligands
a. GSK-3β with Memantine
b. GSK-3β with Alsterpaullone
c. GSK-3β with Celastrol
d. GSK-3β with Quercetin
e. GSK-3β with Kennpaulone
f. GSK-3β with Tideglusib
g. GSK-3β with Withanolide-A
Table 2: List of ligands with their molecular formula and residues involved in Hydrogen bond formation.
| S.No | Ligand names | Molecular Formula | Residues involved in H-bonding |
| 1. | Memantine(reference) | C12H21N | Asp681,Asp700 |
| 2. | Celastrol | C29H38O4 | Phe 567 |
| 3. | Kennpaulone | C16H11BrN2O | Lys 683 |
| 4. | Quercetin | C15H10O7 | Asn 564,Asp 260, Lys 683, Cys 718, Ser 703 |
| 5. | Withanolide A | C28H38O6 | Asp 264, Gln 685,Cys 699,Asn 686 |
| 6. | Alsterpaullone | C16H11N3O3 | Lys 585 |
| 7. | Tideglusib | C19H14N2O2S | 0 |
Table 3: List of the bioactivity score of the ligands.
| S.No | Compound | GPCR ligand | Ion channel modulator | Kinase inhibitor | Nuclear receptor ligand | Protease inhibitor | Enzyme inhibitor |
| 1 | Memantine(reference | -0.28 | 0.12 | -1.09 | -1.07 | -0.60 | -0.47 |
| 2 | Celastrol | -0.07 | -0.22 | -0.26 | 0.58 | -0.03 | 0.63 |
| 3 | Kennpaulone | -0.03 | -0.23 | 0.27 | -0.38 | -0.44 | 0.04 |
| 4 | Quercetin | -0.06 | -0.19 | 0.28 | 0.36 | -0.25 | 0.28 |
| 5 | Withanolide A | 0.04 | 0.32 | -0.43 | 0.71 | 0.15 | 0.86 |
| 6 | Alsterpaullone | 0.03 | -0.12 | 0.22 | -0.20 | -0.30 | 0.04 |
| 7 | Tideglusib | 0.04 | -0.24 | 0.30 | -0.12 | -0.28 | 0.04 |
Table 4: List of Physicochemical properties of the ligands (Lipinski’s Parameters)
| S.No | Compound | Topological polar surface area (Å)2 (TPSA)1 | MW | mi LogP2 | Hydrogen bond donors (nOHNH) | Hydrogen bond acceptors (nON) | Number of rotatable bonds | Lipinski’s violations | % Absorption3 |
| 1 | Memantine(reference | 26.02 | <500 | 2.77 | 2 | 1 | 0 | 0 | 100.0231 |
| 2 | Celastrol | 74.60 | <500 | 5.06 | 2 | 4 | 1 | 1 | 83.263 |
| 3 | Kennpaulone | 44.89 | <500 | 3.72 | 2 | 3 | 0 | 0 | 93.51295 |
| 4 | Quercetin | 131.35 | <500 | 1.68 | 5 | 7 | 1 | 0 | 63.68425 |
| 5 | Withanolide A | 96.36 | <500 | 4.15 | 2 | 6 | 2 | 0 | 75.7558 |
| 6 | Alsterpaullone | 90.71 | <500 | 2.87 | 2 | 6 | 1 | 0 | 77.70505 |
| 7 | Tideglusib | 44.01 | <500 | 3.80 | 0 | 4 | 3 | 0 | 93.81655 |
Topological polar surface area (TPSA) is surface sum of all polar atoms such as oxygen, nitrogen as well as their attatched hydrogen atoms.
LogP is Octanol-water partition coefficient.
Percentage absorption was calculated using formula, % Absorption = 109 − [0.345 × Topological Polar Surface Area].
Conclusions
During the last three decades, the field of molecular docking has emanated for structural based drug designing. Automated docking is widely used for the prediction of biomolecular complexes, in structure and functional analysis and in computer-aided drug designing. It has become an indispensable part of drug discovery and advancement which is utilized for the accurate prediction of protein-ligand complexes. To explore robust and effective medicaments for AD therapy, different phytocompounds were compared against the standard using Autodock 4. The desired ligands were imbeded into the catalytic site of the receptor GSK-3 β and analyzed for the effective protein-ligand interactions. Withanolide-A has shown better results than Memantine wrt. binding affinity and receptor-quercitine complex has highest number of hydrogen bonds i.e. 6 .Therefore, molecular docking identified many more promising, efficacious, selective new drugs in form of Withanolide-A and Quercitine against the Alzheimer’s, reducing the time span of drug discovery process. Apropriate in vitro studies such as ADMET analysis which testifies absorption, distribution, metabolism, and excretion of drugs within the living organism may also be considered further as a lead in drug discovery process.
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Acknowledgements
The authors are thankful to the Department of Biotechnology, University Institute of Engineering and Technology, Kurukshetra (U.I.E.T) for providing the space and resources for this work.
References
- Avila J. Tau phosphorylation and aggregation in Alzheimer’s disease pathology. FEBS Lett. 2006;580:2922–2927. https://doi.org/10.1016/j.febslet.2006.02.067.
CrossRef - Alvarez A.C, Costa A.M Vilarrasa J. The Performance of Several Docking Programs at Reproducing Protein–Macrolide-Like Crystal Structures. Molecules. 2017;22(1):36. DOI:10.3390/molecules22010136.
CrossRef - Brunden K.R, Trojanowski J.Q, Le V.M.Y. Advances in Tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat Rev Drug Discov. 2009;8(10):783–793. https://doi.org/ 1038/nrd2959.
CrossRef - Congdon E.E, Sigurdsson E.M. Tau-targeting therapies for Alzheimer disease. Nature Reviews Neurology. 2018.
CrossRef - Collignon B, Schulz R, Smith J.C, Baudry J. Task-parallel message passing interface implementation of Autodock4 for docking of very large databases of compounds using high-performance super-computers. J Comp Chem. 2011;32:1202–1209. https://doi.org/abs/10.1002/jcc.21696.
CrossRef - Dixit R, Ross J.L, Goldman Y.E, Holzbaur E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. https://doi.org/10.1126/science.1152993.
CrossRef - Filipa I.B, Henriques A.G, Silva A.M.S, Wiltfang J, Silva O.A.B.C. Flavonoids as Therapeutic Compounds Targeting Key Proteins Involved in Alzheimer’s Disease. ACS Chem. Neurosci. 2014;5:83−9292. https://doi.org /1021/cn400213r.
CrossRef - Folch J, Petrov D, Ettcheto M, Abad S, Lopez E.S, Garcia M.L, Olloquequi J, Zarate C.B, Auladell C, Camins A. Current Research Therapeutic Strategies for Alzheimer’s Disease Treatment. Neural Plasticity. 2016;2016: 1-15. https://dx.doi/10.115/2016/8501693.
- Guo T, Noble W, Hanger D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133(5):665–704. DOI: 1007/s00401-017-1707-9.
- Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J Neurochem. 2008; 104(6):1433-1439. https://doi.org/10.1111/j.1471-4159.2007.05194.x.
CrossRef - Islam M.R, Zaman A, Jahan I, Chakravorty R. In silico QSAR analysis of quercetin reveals its potential as therapeutic drug for Alzheimer’s disease. J Young Pharmacists. 2013;5:173-179.
https://doi.org/1016/j.jyp.2013.11.005.
CrossRef - Iqbal K, Liu F, Gong C.X, Iqbal I.G. Tau in Alzheimer Disease and Related Tauopathies. Alzheimer Res. 2010; 7(8):656-664.
CrossRef - Kametani F, Hasegawa M. Reconsideration of amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Neurosci. 2018;12:25. DOI: 10.3389/fnins.2018.00025.
CrossRef - Khan S.A, Khan S.B, Shah Z, Asiri A.M. Withanolides: Biologically Active Constituents in the Treatment of Alzheimer’s Disease. Med Chem. 2016;12(3):238-56. https://doi.org/10.2174/1573406411666151030112314.
CrossRef - Khan, T., Dixit, S., Ahmad, R., Raza, S., Iqbal, A., Joshi, S., Khan, A.R. Molecular docking, PASS analysis, bioactivity score prediction, synthesis, characterization and biological activity evaluation of a functionalized 2-butanone thiosemicarbazone ligand and its complexes. J Chem Biol., 2017; DOI 10.1007/s12154-017-0167-y.
CrossRef - Madeswaran, A., Muthuswamy, U., Kuppusamy, A., Thirumalaisamy, S., Varadharajan, S., Puliyath, J. Docking studies; in silico lipoxygenase inhibitory activity of some commercially available flavonoids. Comput. Method. Mol. Design., 2011; 1 (4):65-72. https:// doi.org/10.3329/bjp.v6i2.9408.
- Madeswaran, A, Muthuswamy, U, Kuppusamy, A, Thirumalaisamy, S, Varadharajan, S, Puliyath, J. Computational drug discovery of potential TAU protein kinase I inhibitors using in silico docking studies. Bangladesh J Pharmacol, 2013; 8: 131-135. https://doi.org/10.3329/bjp.v8i2.13886.
CrossRef - Mahrous, R.S., Ghareeb, D.A., Fathy, H.M., ABU EL-Khair, R.M., Omar, A.A. The Protective Effect of Egyptian Withania somnifera Against Alzheimer’s. Aromat Plants (Los Angels), 2017; 6:2. https://doi.org/10.4172/2167-0412.1000285.
- Martin, L.,Latyopova,X.,Wilson,C.M.,Magnaudix,A.,Prrin,M.L.,Yardin,C., et al. Tau protein kinases; involvement in Alzheimer’s disease. Aging Res Rev 2013;pp 12289-30910, https://doi.org/10.1016/j.arr.2012.06.003.
- Martin, L., Latypova, X., Terro, F., Post-translational modifications of tau protein: implications for Alzheimer’s disease. Int., 2011; 58: 458–471. https://doi.org/ 10.1016/j.neuint.2010.12.06.023.
- Medina, M., Wandosell, F. Deconstructing GSK-3: The Fine Regulation of Its Activity. International Journal of Alzheimer’s Disease., 2011; 2011:1-12. http://dx.doi.org/10.4061/2011/479249.
- Medina M. An Overview on the Clinical Development of Tau-Based Therapeutics . J. Mol. Sci., 2018; 19, 1160 Available from http://www.mdpi.com/journal/ijms DOI: 10.3390/ijms19 041160.
- Motiejunas, D., Wade, R.C. Computer –Assisted Drug Design. Comprehensive Medicinal Chemistry., 2007; 4:193-213.
CrossRef - Pei, J.J., Tanaka, T., Tung, Y.C., Braak, E., Iqbal, K., Grundke-Iqbal, I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J Neuropathol Exp Neurol., 1997; 56(1):70-78.
CrossRef - Schneider, A., Mandelkow, E. Tau-Based Treatment Strategies in Neurodegenerative Diseases. Neurotherapeutics: The J American Society for Experimental NeuroTherapeutics., 2008; 5:443-457. https://org/10.1016/j.nurt.2008.05.006.
- Singh, N., Bhalla, M., Jager, P.D., Gilca, M. An Overview on Ashwagandha: A Rasayana (Rejuvenator) of Ayurveda. African Journal of Traditional, Complementary and Alternative Medicines, 2011; 8:5S.
https://doi.org/10.4314/ajtcam.v8i5S.9.
CrossRef - Spencer, J.P.E. The impact of fruit flavonoids on memory and cognition. British J of Nutrition., 2010; 104:S40–S47. https://doi.org/10.1017/S0007114510003934.
CrossRef - Underwood, E. Tau protein—not amyloid—may be key driver of Alzheimer’s symptoms. Science 2016;https://doi.org/10.1126/science .aaf9985.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.



