

Manuscript accepted on : 25 June 2018
Published online on: 27-06-2018
Plagiarism Check: Yes
Bioinformatics Insights Into Microbial Xylanase Protein Sequences
Deepsikha Anand, Jeya Nasim, Sangeeta Yadav and Dinesh Yadav
Department of Biotechnology, DDU Gorakhpur University, Gorakhpur (U.P) 273009, India.
Corresponding Author E-mail: dinesh_yad@rediffmail.com
DOI : http://dx.doi.org/10.13005/bbra/2631

ABSTRACT: Microbial xylanases represents an industrially important group of enzymes associated with hydrolysis of xylan, a major hemicellulosic component of plant cell walls. A total of 122 protein sequences comprising of 58 fungal, 25 bacterial, 19actinomycetes and 20 yeasts xylanaseswere retrieved from NCBI, GenBank databases. These sequences were in-silico characterized for homology,sequence alignment, phylogenetic tree construction, motif assessment and physio-chemical attributes. The amino acid residues ranged from 188 to 362, molecular weights were in the range of 20.3 to 39.7 kDa and pI ranged from 3.93 to 9.69. The aliphatic index revealed comparatively less thermostability and negative GRAVY indicated that xylanasesarehydrophilicirrespective of the source organisms.Several conserved amino acid residues associated with catalytic domain of the enzyme were observed while different microbial sources also revealed few conserved amino acid residues. The comprehensive phylogenetic tree indicatedsevenorganismsspecific,distinct major clusters,designated as A, B, C, D, E, F and G. The MEME based analysis of 10 motifs indicated predominance of motifs specific to GH11 family and one of the motif designated as motif 3 with sequence GTVTSDGGTYDIYTTTRTNAP was found to be present in most of the xylanases irrespective of the sources.Sequence analysis of microbial xylanases provides an opportunity to develop strategies for molecular cloning and expression of xylanase genes and also foridentifying sites for genetic manipulation for developing novel xylanases with desired features as per industrial needs.
KEYWORDS: Bioinformatics; Multiple sequence Alignment; Source Organisms; Xylanases; Phylogenetic Tree
Download this article as:| Copy the following to cite this article: Anand D, Nasim J, Yadav S, Yadav D. Bioinformatics Insights Into Microbial Xylanase Protein Sequences. Biosci Biotech Res Asia 2018;15(2). |
| Copy the following to cite this URL: Anand D, Nasim J, Yadav S, Yadav D. Bioinformatics Insights Into Microbial Xylanase Protein Sequences. Biosci Biotech Res Asia 2018;15(2). Available from: https://www.biotech-asia.org/?p=30199 |
Introduction
Plant cell wall comprises of three major constituent namely cellulose, hemicelluloses and lignin.Xylan is the major hemicellulosic component and is the second most abundant polysaccharides in nature.Hemicellulose is a branched heteropolymer consisting of pentose and hexose sugars with xylose being most abundant1 (Kumar et al., 2008). A repertoire of enzymes including endo-xylanase (endo-1,4-β-xylanase; E.C.3.2.1.8), β-xylosidase (xylan-1,4-β-xylosidase; E.C.3.2.1.37), α-glucosiduronase (E.C.3.2.1.139),α-arabinofuranosidase(E.C.3.2.1.55) and acetylxylan esterase, (E.C.3.1.1.72)are associated with complete hydrolysis of hemicellulose2(Juturu and Wu, 2012). The endo-xylanase and β-xylosidase are key enzymes associated with hydrolysis of xylan and are collectively referred as xylanases.
Xylanasesbelongs to enzyme class of glycoside hydrolases (GH), which are classified into several families based on amino acid sequences. Xylanases is predominantly represented in two families GH10 and GH113-5 (Paes et al., 2012;Lafond et al., 2014; Chakdar et al., 2016).Xylanases have been reported from diverse microbial sources namely fungi, bacteria,actinomycetes and yeast and have been reviewed extensively over the years2,6-10 (Beg et al., 2001; Collins et al., 2005; Nair et al., 2008; Juturu and Wu, 2012; Juturu and Wu, 2014; Walia et al., 2017).Xylanasesrepresents industrially important group of enzymes with diverse applications like pulp and paper bleaching, fruit juice clarification, bioethanol production, bioconversion etc.2,11-14.(Shatalov et al., 2008; Valls et al., 2010; Juturu and Wu, 2012; Singh et al., 2013; Walia et al., 2015).
Several bioinformatics studies have been done on various xylanases. In-silico analysis of structural attributes ofcommercially important xylanases from diverse sources structural has beenreported15(Arora et al., 2009).Attempts have been made to study the structural dynamics changes of the Trichodermalongibrachiatumxylanase upon binding with xylohexaose and xylan ligands16 (Uzuner et al., 2010). In-silico structural prediction of Bacillus brevisxylanase and its comparative assessment with few bacterial and fungal xylanases has been reported recently17(Mathur et al., 2015).Efforts have been made to analyze several plant cell wall degrading enzymes (PCWDEs) including xylanses and polygalacturonasesofFusariumvirguliformeusing bioinformatics tools to develop fungal resistant soyabean18(Chang et al., 2016). Homology modeling of xylanase from Aspergillusfumigatus R1 isolate to get an insight into three dimensional structurehas been attempted19 (Deshmukh et al., 2016).
This manuscript reports in-silico characterization of xylanase protein sequences retrieved from NCBI representing diverse microbial sources namely fungi, bacteria, actinomycetes and yeast. Bioinformatics assessment of these sequences for homology,sequence alignment, physio-chemical attributes, motif assessment and phylogenetic tree construction isreported.The bioinformatics driven characterization of available sequences of microbial xylanases could be utilized for developing appropriate strategies for molecular cloning and expression of xylanasegenes.Further, the sequence-structure-function relationship could be established from in-silico studies and novel xylanases could be derived using state-of-the art technologies either metagenomics or directed evolution approaches.
Materials and Methods
Database Search and Sequence Retrieval
Xylanase protein sequences representing different microbial sources were retrieved from GenBank, NCBI (http//www.ncbi.nlm.nih.gov/).The sequences retrieved were saved in FASTA format and truncated proteins were discarded. The major groups as source organisms represents fungi, bacteria, actinomycetes and yeast and the all the sequences of xylanases belongs to GH11 family.
Physio-Chemical Attributes
The physio-chemical attributes namely molecular weight, theoretical pI, aliphatic index, instability index, Grand Average of Hydropathicity (GRAVY) were analyzed by ProtParam tool (http://web.expasy.org/protparam/).20(Gasteiger et al.,2005).
Multiple sequence Alignment and Phylogenetic Analysis
The protein alignment of full length amino acid sequences of xylanase were performed by CLUSTAL X version 2.121(Larkin et al., 2007). Phylogenetic tree was constructed by NJ method using the MEGA 7.0 program22(Kumar et al., 2016) based on protein sequences.
Identification of conserved motifs
The protein sequences of xylanase were analyzed by Multiple EM for Motif Elicitation(MEME)program version 4.12.0 (http://meme.nbcr.net/meme/)23(Timothy et al.,2009).The maximum number of motifswereset as 10.The minimum width of 6 and maximum width of 50 amino acids was set along with other factors as default values.
Results and Discussion
Physio-chemical characterization of xylanases
A total of the 122 xylanase protein sequences belonging to GH11 family representing 58 fungal, 25 bacterial, 19 actinomycetes and 20 yeastxylanaseswere retrieved from NCBI databases (Table-1).It has been reported that bacterial xylanases generally represent GH10 family though fungal xylanases predominantly belongs to GH11 family24(Liu et al., 2011).Theirphysio-chemical properties namely molecular weight, pI, instability index, aliphatic index, GRAVYwere analyzed using ProtParam tool (Table-1). Theamino acid residues ranged from 188-362 residueswhilemolecularweightwasin the range of 20.3-39.7 KDa. The Isoelectric point(pI)was in the range of 3.93-9.69.The molecular weight in the range of 8.5 to 85kDa and pI in the range of 4-10.3 has been reported for bacterial5(Chakdhar et al.,2015) and fungal xylanases25(Polizeli et al. 2005).
Table 1: List of xylanase protein sequences from different microbial sources with in-silicophysio-chemical attributes revealed by Protparam
| S.No. | Source Organism | Accession No. | Amino acid | Mol.wt. | pI | Instability
index |
Aliphatic
index |
GRAVY |
| FUNGI | ||||||||
| 1 | Aspergillusnomius | XP_015411959 | 229 | 24.4 | 5.73 | 26.21 | 59.61 | -0.33 |
| 2 | Aspergillusnomius | XP_015411268 | 221 | 23.8 | 4.55 | 22.18 | 60.9 | -0.427 |
| 3 | Aspergillusniger | AAS46914 | 225 | 24.1 | 5.23 | 22.11 | 59.78 | -0.396 |
| 4 | Aspergillusniger | AAS46913 | 211 | 22.5 | 4 | 19.86 | 59.62 | -0.178 |
| 5 | Aspergillusniger | AAA99065 | 211 | 22.7 | 4.55 | 26.49 | 59.15 | -0.18 |
| 6 | Aspergillusniger | AFK10491 | 225 | 24 | 5.2 | 21.06 | 58.53 | -0.38 |
| 7 | Aspergillusniger | CAA03654 | 225 | 24 | 5.45 | 21.06 | 58.53 | -0.384 |
| 8 | Aspergillusniger | ACN89393 | 225 | 24 | 5.2 | 21.06 | 58.53 | -0.38 |
| 9 | Aspergillusniger | AAM95167 | 225 | 24.1 | 5.23 | 22.11 | 59.78 | -0.396 |
| 10 | Aspergillusniger | ABA00146 | 225 | 24.1 | 5.23 | 22.11 | 59.78 | -0.396 |
| 11 | Aspergillusniger | AGH29125 | 225 | 24.1 | 5.23 | 22.11 | 59.78 | -0.396 |
| 12 | Aspergillusflavus | KOC12560 | 232 | 24.6 | 5.49 | 21.53 | 58.84 | -0.291 |
| 13 | Aspergillusfumigatus | XP_751100 | 221 | 23.8 | 5.22 | 24.15 | 56.02 | -0.422 |
| 14 | Aspergillusfumigatus | XP_748354 | 228 | 24.4 | 6.27 | 23.44 | 53.9 | -0.361 |
| 15 | Aspergillusfumigatus | XP_748367 | 313 | 33 | 6.03 | 26.03 | 69.71 | -0.093 |
| 16 | Aspergillusnidulans | CAA90074 | 221 | 23.5 | 4.54 | 32.04 | 54.71 | -0.378 |
| 17 | Aspergillusniger | XP_001388522 | 225 | 24 | 5.2 | 21.06 | 58.53 | -0.38 |
| 18 | Aspergillusniger | ACJ26382 | 225 | 24 | 5.2 | 21.06 | 58.53 | -0.384 |
| 19 | Aspergillusniger | ACA24724 | 225 | 24 | 5.44 | 20.72 | 59.82 | -0.346 |
| 20 | Aspergillusniger | AAM08362 | 225 | 24.1 | 5.23 | 22.11 | 59.78 | -0.396 |
| 21 | Aspergillusniger | XP_001389848 | 231 | 24.8 | 3.94 | 26.61 | 61.69 | -0.364 |
| 22 | Aspergillusniger | GAQ35804 | 231 | 24.8 | 3.93 | 25.84 | 61.26 | -0.373 |
| 23 | Aspergillusniger | GAQ46944 | 211 | 22.5 | 4.07 | 17.72 | 60.52 | -0.138 |
| 24 | Aspergillusniger | EHA24718 | 236 | 25.7 | 4.4 | 21.78 | 65.64 | -0.408 |
| 25 | Aspergillusniger | ALN49265 | 256 | 28 | 4.7 | 34.64 | 70.04 | -0.312 |
| 26 | Aspergillusniger | AFK10490 | 211 | 22.5 | 4.31 | 23.47 | 58.72 | -0.146 |
| 27 | Aspergillusniger | ADO66655 | 211 | 22.6 | 4.31 | 22.55 | 58.72 | -0.145 |
| 28 | Aspergillusniger000000 | XP_001401361 | 211 | 22.6 | 4.31 | 23.06 | 58.72 | -0.156 |
| 29 | Aspergillusniger | ACN82438 | 211 | 22.5 | 4.39 | 23.33 | 57.35 | -0.17 |
| 30 | Aspergillusniger | GAQ40597 | 256 | 28 | 4.76 | 34.57 | 70.78 | -0.303 |
| 31 | Aspergillusoryzae | XP_001823798 | 232 | 24.4 | 5.48 | 21.33 | 59.52 | -0.276 |
| 32 | Aspergillusoryzae | XP_001818666 | 221 | 23.7 | 4.67 | 19.63 | 61.76 | -0.424 |
| 33 | Aspergillusclavatus | XP_001273882 | 192 | 20.4 | 9.63 | 32.86 | 57.92 | -0.357 |
| 34 | Aspergillusluchuensis | GAT31123 | 225 | 24.1 | 5.74 | 22.92 | 60.22 | -0.432 |
| 35 | Aspergillusluchuensis | GAT25039 | 211 | 22.6 | 4.07 | 20.58 | 59.15 | -0.129 |
| 36 | Aspergillusluchuensis | OJZ80582 | 211 | 22.6 | 4.02 | 21.52 | 57.77 | -0.153 |
| 37 | Aspergillusluchuensis | GAT30893 | 256 | 28 | 4.76 | 34.57 | 69.26 | -0.304 |
| 38 | Aspergillusluchuensis | OJZ85846 | 256 | 28 | 4.84 | 35.16 | 68.87 | -0.308 |
| 39 | Aspergillusversicolor | ABM55503 | 206 | 21.9 | 4.24 | 16.32 | 62.96 | -0.065 |
| 40 | Fusariumavenaceum | KIL90649 | 287 | 29.8 | 4.53 | 29.56 | 42.93 | -0.625 |
| 41 | Fusariumverticillioides | XP_018753195 | 314 | 32.6 | 4.64 | 24.61 | 37.96 | -0.832 |
| 42 | Fusariumverticillioides | XP_018753196 | 296 | 30.6 | 4.87 | 21.85 | 39.93 | -0.749 |
| 43 | Fusariumverticillioides | XP_018755440 | 233 | 25.2 | 8.98 | 30.08 | 58.93 | -0.436 |
| 44 | Fusariumverticillioides | XP_018761356 | 231 | 25.7 | 6.41 | 34.6 | 51.95 | -0.694 |
| 45 | Fusariummangiferae | CVK98696.1 | 231 | 25.6 | 6.41 | 34.56 | 51.52 | -0.685 |
| 46 | Fusariumproliferatum | CVL11933 | 231 | 25.6 | 6.05 | 36.73 | 53.2 | -0.646 |
| 47 | Fusariumfujikuroi | CCT69575 | 231 | 25.7 | 6.41 | 36.04 | 53.2 | -0.662 |
| 48 | Fusariumpseudograminearum | XP_009253878 | 231 | 25.7 | 6.5 | 32.22 | 52.79 | -0.666 |
| 49 | Fusariumlangsethiae | KPA38457.1 | 228 | 24.5 | 9.17 | 26.82 | 58.64 | -0.472 |
| 50 | Fusariumoxysporum | EWZ00952 | 277 | 29.2 | 4.91 | 23.97 | 41.59 | -0.736 |
| 51 | Fusariumoxysporum | EWZ46984 | 289 | 30.3 | 4.83 | 24.9 | 41.87 | -0.754 |
| 52 | Fusariumoxysporum f. sp.vasinfectum | EXM22239 | 271 | 28.3 | 5.19 | 22.81 | 43.21 | -0.676 |
| 53 | Fusariumoxysporum f. sp.vasinfectum | EXM22238 | 289 | 30.3 | 4.83 | 25.75 | 40.87 | -0.77 |
| 54 | Fusariumoxysporum f. sp. lycopersici | XP_018238679 | 277 | 28.8 | 5.06 | 23.67 | 42.64 | -0.686 |
| 55 | Fusariumoxysporum f. sp. lycopersici | XP_018238678 | 286 | 29.7 | 4.98 | 24.79 | 40.63 | -0.737 |
| 56 | Fusariumoxysporum f. sp. lycopersici | XP_018246999 | 232 | 25.1 | 8.96 | 25.04 | 57.46 | -0.479 |
| 57 | Fusariumoxysporum f. sp. lycopersici | XP_018256151 | 231 | 25.6 | 6.18 | 32.52 | 52.77 | -0.655 |
| 58 | Fusariumoxysporum f. sp. cubense | EMT73821 | 231 | 25.6 | 6.41 | 34.17 | 54.46 | -0.638 |
| BACTERIA | ||||||||
| 59 | Bacillus cereus | AAZ17391 | 213 | 23.3 | 9.44 | 15.87 | 54.46 | -0.425 |
| 60 | Dictyoglomusthermophilum | WP_012547705 | 360 | 39.7 | 8.68 | 25.12 | 72.28 | -0.314 |
| 61 | Dictyoglomusthermophilum | AAC46361 | 360 | 39.7 | 8.68 | 19.75 | 71.19 | -0.333 |
| 62 | Dictyoglomusturgidum | WP_012582654 | 356 | 39.4 | 8.45 | 22.94 | 71.99 | -0.285 |
| 63 | Fibrobactersuccinogenes | WP_014546846 | 327 | 36.1 | 5.07 | 19.20 | 65.32 | -0.350 |
| 64 | Paenibacillusjilunlii | WP_062524300 | 212 | 23.2 | 9.10 | 24.53 | 56.08 | -0.342 |
| 65 | Paenibacilluspolymyxa | ADK47978 | 211 | 22.7 | 9.55 | 17.82 | 61.47 | -0.303 |
| 66 | Paenibacilluspolymyxa | KOS03251 | 212 | 23.2 | 9.40 | 21.41 | 51.93 | -0.407 |
| 67 | Paenibacilluspolymyxa | WP_025720875 | 212 | 23.1 | 9.40 | 21.41 | 51.93 | -0.419 |
| 68 | Paenibacilluspolymyxa | WP_061831741 | 212 | 23.1 | 9.15 | 21.05 | 51.93 | -0.417 |
| 69 | Paenibacilluspolymyxa | WP_013308993 | 212 | 23.2 | 9.15 | 20.83 | 51.93 | -0.432 |
| 70 | Paenibacilluspolymyxa | WP_016820426 | 212 | 23.1 | 9.30 | 22.35 | 55.14 | -0.409 |
| 71 | Paenibacilluspolymyxa | WP_017425612 | 212 | 23.2 | 9.30 | 22.05 | 53.30 | -0.417 |
| 72 | Paenibacilluspolymyxa | WP_023987219 | 212 | 23.1 | 8.94 | 20.56 | 51.93 | -0.414 |
| 73 | Paenibacilluspolymyxa | WP_031462284 | 212 | 23.1 | 9.30 | 21.95 | 55.14 | -0.407 |
| 74 | Paenibacilluspolymyxa | WP_013373220 | 212 | 23.2 | 9.40 | 21.91 | 53.77 | -0.407 |
| 75 | Paenibacilluspolymyxa | WP_058831015 | 212 | 23.1 | 9.40 | 19.84 | 55.61 | -0.394 |
| 76 | Paenibacilluspolymyxa | WP_039272535 | 212 | 23.1 | 9.18 | 23.35 | 52.41 | -0.404 |
| 77 | Paenibacilluspolymyxa | WP_064797296 | 212 | 23.1 | 9.40 | 16.99 | 53.77 | -0.445 |
| 78 | Paenibacilluspolymyxa | WP_023987332 | 362 | 39.5 | 7.69 | 26.27 | 59.78 | -0.524 |
| 79 | Paenibacilluspolymyxa | WP_068938485 | 362 | 39.5 | 7.69 | 25.82 | 59.50 | -0.525 |
| 80 | Paenibacilluspolymyxa | WP_071639791 | 362 | 39.5 | 7.69 | 19.53 | 62.18 | -0.504 |
| 81 | Paenibacillusriograndensis | WP_020430448 | 212 | 23.1 | 9.25 | 20.64 | 56.56 | -0.316 |
| 82 | Paenibacillusriograndensis | WP_060864761 | 212 | 23.2 | 9.25 | 20.73 | 56.08 | -0.320 |
| 83 | Paenibacillus terrae | WP_014280040 | 212 | 23.1 | 9.30 | 21.81 | 52.41 | -0.405 |
| ACTINOMYCETES | ||||||||
| 84 | Actinobacteria | WP_054228265 | 330 | 35.0 | 9.17 | 28.72 | 51.42 | -0.415 |
| 85 | Hamadaeatsunoensis | WP_027341164 | 327 | 33.8 | 9.16 | 26.33 | 56.70 | -0.309 |
| 86 | Herbidosporacretacea | WP_061296570 | 321 | 34.0 | 9.49 | 29.13 | 50.16 | -0.513 |
| 87 | Herbidosporamongoliensis | WP_066360436 | 322 | 33.8 | 9.37 | 25.86 | 48.79 | -0.493 |
| 88 | Microbispora sp. | WP_055478269 | 335 | 35.5 | 9.69 | 32.99 | 49.55 | -0.569 |
| 89 | Micromonosporacoxensis | SCG34253 | 330 | 34.5 | 9.42 | 33.72 | 53.82 | -0.366 |
| 90 | Micromonosporanigra | SCL32394 | 329 | 34.5 | 9.61 | 32.39 | 54.83 | -0.395 |
| 91 | Nocardiopsisdassonvillei | WP_061080181 | 332 | 35.2 | 8.74 | 35.03 | 51.99 | -0.497 |
| 92 | Nonomuraeajiangxiensis | SDH13383 | 321 | 33.9 | 9.32 | 36.29 | 56.20 | -0.397 |
| 93 | Planomonosporasphaerica | WP_068897915 | 335 | 35.1 | 9.60 | 32.76 | 48.39 | -0.503 |
| 94 | Saccharothrixsyringae | WP_033431747 | 329 | 34.8 | 9.67 | 30.96 | 53.98 | -0.495 |
| 95 | Streptomonospora alba | WP_040275156 | 339 | 35.8 | 5.17 | 35.81 | 44.31 | -0.605 |
| 96 | Streptomyces hirsutus | WP_055594006 | 337 | 35.9 | 9.30 | 26.42 | 52.37 | -0.406 |
| 97 | Streptomyces reticuli | WP_059255807 | 337 | 35.8 | 9.53 | 29.68 | 54.69 | -0.413 |
| 98 | Streptomyces viridosporus | AAF09501 | 329 | 35.1 | 9.55 | 26.02 | 50.09 | -0.518 |
| 99 | Streptomyces davawensis | WP_015659056 | 320 | 33.8 | 9.23 | 23.06 | 56.38 | -0.351 |
| 100 | Streptomyces aureus | WP_051901343 | 313 | 32.6 | 8.95 | 20.17 | 52.36 | -0.394 |
| 101 | Thermobifidafusca | WP_011291660 | 338 | 36.4 | 9.47 | 34.31 | 52.28 | -0.495 |
| 102 | Thermobifidafusca | WP_016188539 | 338 | 36.4 | 9.37 | 34.20 | 52.28 | -0.504 |
| YEAST | ||||||||
| 103 | Aureobasidiummelanogenum | KEQ63689 | 218 | 23.3 | 4.73 | 30.22 | 70.78 | -0.204 |
| 104 | Aureobasidiummelanogenum | KEQ63789 | 217 | 23.4 | 8.27 | 20.93 | 52.63 | -0.369 |
| 105 | Aureobasidiummelanogenum | KEQ64351 | 221 | 23.4 | 4.86 | 17.74 | 60.90 | -0.108 |
| 106 | Aureobasidiummelanogenum | BAB69655 | 221 | 23.3 | 4.86 | 17.74 | 61.36 | -0.096 |
| 107 | Aureobasidiumnamibiae | XP_013425857 | 225 | 24.1 | 9.25 | 24.83 | 60.71 | -0.432 |
| 108 | Aureobasidiumnamibiae | XP_013422490 | 218 | 23.1 | 6.40 | 25.16 | 72.11 | -0.204 |
| 109 | Aureobasidiumnamibiae | XP_013429521 | 221 | 23.2 | 5.71 | 18.17 | 62.26 | -0.138 |
| 110 | Aureobasidiumpullulans | KEQ83780 | 229 | 25.0 | 8.84 | 22.97 | 48.52 | -0.514 |
| 111 | Aureobasidiumpullulans | KEQ90048 | 224 | 24.1 | 9.03 | 24.80 | 58.39 | -0.376 |
| 112 | Aureobasidiumpullulans | KEQ80629 | 218 | 23.2 | 5.54 | 35.83 | 75.23 | -0.176 |
| 113 | Aureobasidiumpullulans | AAD51950 | 221 | 23.5 | 5.29 | 16.84 | 58.73 | -0.181 |
| 114 | Aureobasidiumsubglaciale | XP_013347844 | 224 | 24.1 | 8.84 | 29.17 | 58.35 | -0.420 |
| 115 | Aureobasidiumsubglaciale | XP_013339677 | 230 | 25.0 | 7.77 | 34.07 | 52.57 | -0.464 |
| 116 | Baudoiniapanamericana | XP_007672582 | 188 | 20.3 | 6.54 | 23.04 | 61.22 | -0.298 |
| 117 | Bispora sp. | ADZ99365 | 205 | 21.8 | 4.21 | 14.67 | 59.41 | -0.281 |
| 118 | Cryptococcus sp. | BAA09699 | 209 | 22.7 | 5.46 | 19.73 | 50.81 | -0.515 |
| 119 | Cryptococcus sp. | BAA09698 | 209 | 22.7 | 5.46 | 19.73 | 50.81 | -0.515 |
| 120 | Pseudozymahubeiensis | XP_012187186 | 261 | 27.9 | 9.15 | 24.47 | 50.84 | -0.466 |
| 121 | Saitozymaflava | AOS95422 | 209 | 22.7 | 6.25 | 18.09 | 51.29 | -0.499 |
| 122 | Saitozymaflava | ABY50453 | 209 | 22.7 | 6.25 | 21.81 | 52.20 | -0.506 |
The stability –instability index method26(Guruprasad et al. 1990) estimates the stability of the protein in a test tube. The instability index less than 40 predicts a stable protein whereas values higher than 40 denotes potentially unstable protein. The value of instability index of xylanases ranged from 14.67-36.73, which is less than 40 and hence represents stable protein.The stability -aliphatic index method20(Gasteiger et al., 2005) reflects regional stability based on the relative volume occupied by aliphatic side chain and is a positive indicator of globular protein theromostability.The aliphatic index of xylanaseis the range of 37.96-75.23 as reported in literature27(Walia et al., 2015) and high aliphatic index indicates stability of xylanases for wide temperature range. The aliphatic index of xylanases protein sequences from Aspergillusniger (ALN49265), Dictyoglomusthermophilum(WP_012582654, AAC46361), Aureobasidiummelanogenum (KEQ63689), Aureobasidiumnamibiae (XP_013422490) and Aureobasidiumpullulans(KEQ80629) was above 70 (Table-1).Another important physio-chemical attributeanalyzed by ProtParam is GRAVY value derived by calculating the sum of hydropathy values28(Kyte and Doolittle, 1982) of all the amino acids, divided by the number of residues in the sequence20(Gasteiger et al. 2005). Increasing positive score indicates a greater hydrophobicity. The microbial xylanase protein sequences revealed negative GRAVY value ranging from -0.832 to -0.093 indicating hydrophilic nature.
Multiple Sequence Alignment Analysis
Multiple sequence alignment ofretrievedxylanasesequenceswasperformed by CLUSTAL X version 2.1andis shown in Figure 1(A,B,C& D). Several conserved amino residues are observed for different source organisms while comprehensive multiple sequence alignment of all122xylanase sequences revealedtwohighlyconserved residues namely YGW and EYYI (Figure-1E). The presence of these conserved amino acid residues has been reported for xylanases especially from fungal and bacterial sources29-31(Ellouze et al.,2011, Sapag et al., 2002,Torronen et al.,1992).Similar conserved amino acid residues have been observed for xylanaseofT. longibrachiatum. Another conserved amino acid residues with sequence RVNEPSIQGTATFNQYhas been reported, which plays significant role in stabilizing during substrate binding16(Uzuner et al.,2010).
 |
Figure 1: Multiple sequence alignment of xylanase protein sequences from (A) Fungal (B) Bacterial (C) Actinomycetes and (D) Yeast sources.Strongly conserved amino acid residues are indicated by asterisk* above the alignment. |
 |
Figure 1.e: Combined sequence alignment of a total 122xylanases protein sequences from different microbial sources.Strongly conserved amino acid residues are indicated by asterisk* above the alignment. |
It has also been reported that glutamate amino acid residue responsible for catalysis is conserved in genera of ascomycetes and basidiomycetes representing GH11 and GH10 family of xylanases32(Cervanteset al., 2016).Xylanase proteins representing actinomycetes revealed several conserved amino acid residues at positions 119-131,155-169,185-191,197-208 and 221-233 along with YGW and EYY residues (Figure-1C). Similarly alignment of 20 xylanase protein sequences of GH11 family from source organism yeast revealed several conserved amino acid residues at position 214-216 (Figure-1D). The presence of conserved amino acid residues provides an insight into the catalytic activity of the enzyme based on the fact that there exits sequence-structure-function relationship. The multiple sequence alignment also provides an opportunity to design appropriate degenerate primers for amplification of xylanase genes from different microbial sources.
Phylogenetic Analysis
The phylogenetic tree based on microbial xylanase protein sequenceswere constructed by NJ method (Figure-2 A, B, C, D). The phylogenetic tree representing fungal xylanase protein sequences revealed 5 distinct major clusters designated as I,II,III,IV and V group (Figure-2A).
 |
Figure 2.a: Phylogenetic tree constructed using protein sequences of 58 fungal xylanase. |
The distinct major clusters designated as I, II, III, IV and V comprising of19, 19, 5, 10 and 5 members respectively are highlighted
Genera specific clusters for different species of Aspergillus and Fusarium were observed. This indicates sequence level similarity among xylanases representing specific genera and could be utilized to decipher specific sequence features for designing genera specific probe or primers exclusively for xylanase genes. Further distinct sub-clusters representing multiple strains of predominately Aspergillusniger and Fusariumoxysporum were also observed (Figure-2A). In case of bacterial xylanases two distinct clusters designated as I and II comprising exclusively forPaenibacillusandDictyoglomus species were observed (Figure-2B).
 |
Figure 2.b: Phylogenetic tree constructed using 25 protein sequences of xylanasesfrom bacterial sources. |
The distinct major clusters designated as I and II comprising of 21 and 3 members respectively are highlighted.
Xylanase from Fibrobactersuccinogenes occupied distinct place in the phylogenetic tree. The major clusters I and II represented predominantly multiple strains of Paenibacilluspolymyxa and Dictyoglomusthermophilum indicating strain specific sequence similarity.Similarly, the phylogenetic tree for xylanasesfromactinomycetes revealed two major clusters I and II with 15 and 4 members respectively. The major cluster I was further divided into three subclusters i.e. A, B, C (Figure-2C).
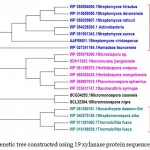 |
Figure 2.c: Phylogenetic tree constructed using 19 xylanaseprotein sequences of actinomycetes. |
The distinct major clusters designated as I and II comprising of 15 and 4 members respectively are highlighted.
In case of xylanases from yeast sources, two major clusters I and II with 12 and 8 sequences were observed, which were further divided into two sub-clusters A and B respectively (Figure-2D).The phylogenetic tree comprising of all the 121 sequences representing different microbial sources revealed seven distinct major clusters designated as A, B, C, D, E, F and G. These major clusters represented specific source organisms (Figure-2D). The major cluster A comprising of 19 sequences represents actinomycetes source organism exclusively while B represented bacterial sources. The major cluster C with 12 sequences represents both bacterial and fungal sources while D comprises of 22 sequences exclusively from Aspergillus genera.
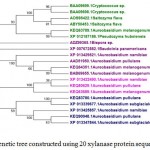 |
Figure 2.d: Phylogenetic tree constructed using 20 xylanase protein sequences of yeast. |
The distinct major clusters designated as I and II comprising of 12 and 8 members respectively are highlighted.
The major cluster E included 3 sequences of yeast genera, F included 21 sequences predominantly from Fusariumgenera along with some sequences from yeast and the major cluster G with 27 sequences comprises of both fungal and yeast source organisms (Figure-2D). Phylogenetic tree revealing xylanases representing GH10 and GH11 family and also basidiomycetes and ascomycetes specific fungal groups have been reported32,29(Cervanteset al.,2016; Ellouzeet al.,2011). Distinct clades representing GH10, GH11 and GH30 family revealing evolutionary relatedness based on 22 protein sequences of xylanaseswerealso deciphered33(Liao et al., 2015).
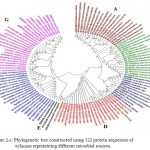 |
Figure 2.e: Phylogenetic tree constructed using 122 protein sequences of xylanase representing different microbial sources. |
The major clusters designated as A,B,C,D,E,F and G is highlighted.
Motif distribution And characterization
The conserved motifs deduced by MEME are generally analyzed for biological function using protein BLAST and domains are characterized by Interproscanto reveal the best possible match based on highest similarity score.The distribution of five motifs among microbial xylanase protein sequencesis shown in Figure 3A, B, C and D.
 |
Figure 3: Distribution of 5 commonly observed motifs among xylanases representing different microbial sources (A) Fungal (B) Bacterial (C) Actinomycetes and (D) Yeast sources. |
The distribution of five motifs among 58 fungal xylanase protein sequences was analyzed (Figure3A) and motifs with width and best possible match amino acid sequences is shown in Table-2A. The predominance of motifs with conserved domain representing unique feature of GH11 family was observed. The motif 1 with amino acid sequence IDGTATFTQYWSVRQNKRSSGTVTTSNHFNAWAKLGMNLGTHNYQIVATE and motif 2 with sequence PSGNGYLSVYGWTTNPLVEYYIVESYGTYNPGSGGTYKGTV was uniformly distributed among fungal xylanases. Similarly the motif assessment for bacterial(Figure-3B, Table-2B), actinomycetes (Figure-3C, Table-2C) and yeast (Figure-3D, Table-2D) source organisms revealed predominance of conserved domains specific to GH11 family.
Table 2.a: The best possible match amino acid sequences of five motif with respective domains observed for xylanasesinFungal, Bacterial, Actinomycetes and Yeast.
| Motif no. | Sequencelength | Sequence | Occurrence at different site | Conserved
Domain |
| (A) FUNGAL | ||||
| 1 | 50 | IDGTATFTQYWSVRQNKRSSGTVTTSNHFNAWAKLGMNLGTHNYQIVATE | 58 | GH11family |
| 2 | 41 | PSGNGYLSVYGWTTNPLVEYYIVESYGTYNPGSGGTYKGTV | 58 | GH11family |
| 3 | 21 | GNFVGGKGWNPGSARTITYSG | 56 | GH11family |
| 4 | 21 | NNGFYYSFWTDGGGDVTYTNG | 47 | GH11family |
| 5 | 15 | DGSTYDIYTTTRTNA | 57 | No information |
| (B) BACTERIAL | ||||
| 1 | 41 | AGVWAPSGNGYLALYGWTRNSLIEYYVVDSWGTYRPTGTYK | 25 | GH11family |
| 2 | 21 | SDGGTYDIYTTMRYBAPSIEG | 24 | GH11family |
| 3 | 29 | ITFSNHVKAWASKGMNLGSNWSYQVLATE | 21 | GH11family |
| 4 | 21 | AATDYWQNWTDGGGTVNAVNG | 21 | No information |
| 5 | 29 | GGNYSVTWKBTGNFVVGKGWTTGSPNRTI | 21 | GH11family |
| (C) ACTINOMYCETES | ||||
| 1 | 50 | YDIYKTTRYNAPSIEGTRTFDQYWSVRQSKRTGGTITSGNHFDAWARAGM | 19 | GH11family |
| 2 | 50 | RRSVTYSGSFNPSGNAYLTLYGWTRNPLVEYYIVDNWGTYRPTGTYKGTV | 19 | GH11family |
| 3 | 50 | CTATLSAGQQWSDRYNLNVSVSGSSNWTVTMNVPSPAKVJSTWNVSASYP | 19 | No information |
| 4 | 50 | VTTNQTGTNNGYFYSFWTDSQGTVSMELGSGGNYSTSWRNTGNFVAGKGW | 18 | GH11family |
| 5 | 29 | LTARPNGNGNNWGVTIQHNGNWTWPTVSC | 19 | No information |
| (D) YEAST | ||||
| 1 | 50 | SDGSTYDVCTDTRTNQPSITGTSTFKQYWSVRQNKRTSGTVTTQNHFNYW | 20 | GH11family |
| 2 | 16 | WTNSPLVEYYVIESYG | 20 | GH11family |
| 3 | 23 | GSYNYQVMATEGFSGSGSASVTV | 19 | GH11family |
| 4 | 21 | NTDFVVGLGWSTGAARTITYS | 20 | GH11family |
| 5 | 29 | INYVQNYNGNVANFTYNZNAGTYSMNWNN | 12 | No information |
The comprehensive analysis of all the xylanases sequences, irrespective of source organisms for motif distribution and domain characterization is shown in Figure-3E and Table-2B respectively. A total of 10 motifs among the 122 xylanase protein sequences revealed 6 motifs with domains specific to GH11 family. The Motif 3 with sequence GTVTSDGGTYDIYTTTRTNAP was found to be highly conserved and was found uniformly among most of the microbial xylanase sequences analyzed. The sequence motifs could be considered as signature sequence revealing the functional identity of the proteins or enzymes and could be targeted for enzyme engineering. The motif assessment also provides an insight into the structural and functional diversity of the enzymesas reported34 (Mohammed et al., 2011). Motif assessment for Endo-1,4-β-xylanase of GH11 family from source organism Paecilomycesvariotii, Schizophyllum commune and Trichodermaharzianumhas been reported15(Arora et al.,2009).
 |
Figure 3.e: Distribution of 10 commonly observed motifs among 122 xylanase protein sequences. |
Table 2.b: The best possible match amino acid sequences of 10 motifs with respective conserved domain observed among 122 protein sequences of xylanases from different microbial sources.
| Motif no. | Sequence length | Sequence | Occurrence at different site | Conserved
Domain |
| 1 | 21 | NSYLAVYGWTRNPLVEYYIVE | 118 | GH 11Family |
| 2 | 18 | IDGTATFTQYWSVRQSKR | 118 | GH 11Family |
| 3 | 21 | GTVTSDGGTYDIYTTTRTNAP | 121 | GH11Family |
| 4 | 17 | TVTTGNHFBAWASLGMN | 120 | GH11Family |
| 5 | 21 | HBYQILATEGYQSSGSSSITV | 120 | GH11Family |
| 6 | 15 | WSNTGNFVGGKGWNT | 115 | No information |
| 7 | 29 | NNGYYYSFWTDGGGTVTYTNGSGGNYSVE | 84 | GH11Family |
| 8 | 50 | CTATLSAGQQWSDRYNLNVSVSGSSNWTVTMNVPSPAKVJSTWNVSASYP | 19 | No information |
| 9 | 15 | SARTITYSGSFNPSG | 120 | No information |
| 10 | 11 | SYGTYNPGSGY | 119 | No information |
The relevance of bioinformaticsin enzyme engineering has been witnessed in recent years and several in-silico tools mainly focusing on prediction of three dimensional structure of enzyme based on the availability of the protein sequences is now being routinely used35,36(Damborsky and Brezovsky, 2014; Suplatov et al., 2015). The in-silico analysis of the sequences of genes/proteins of several industrially important enzymes mainly focusing on homology search, multiple sequence alignment, phylogenetic tree construction and motif assessment has been reported.37-48(Yadavet al., 2009; Dubeyet al., 2010; Yadavet al., 2010; Malviyaet al., 2011; Dubey et al., 2012; Moryaet al., 2012; Yadav et al., 2012; Kumar et al., 2012; Dwivedi and Mishra, 2014; Mathew et al., 2014; Moryaet al., 2016;; Yadavet al., 2017).
Molecular cloning of relevant genes coding for enzymes and its expression needs bioinformatics interventiontargeting forsubstantial improvement in enzyme for desired features.Recently,functional diversity of multiple xylanases from Penicilliumoxalicum GZ-2, revealing functional redundancy using bioinformatics approach has been reported.33(Liao et al., 2015)
Conclusions
Using bioinformatics approach, an attempt has been made to characterize microbial xylanase sequences for several important attributes, which could be targeted for enzyme engineering to develop novel xylanases. The knowledge about the sequences is being applied for deciphering the three dimensional structure using appropriate in-silicotoolsprior to wet-lab experimentation. The tools of bioinformatics are also relevant in the era of genomics, where several microbial genome sequences have been deciphered. This provides an opportunity to perform genome-wide identification and characterization of multigene families of industrially important enzymes and analyze the functional redundancy.There has been substantial improvement in advanced enzyme technologies including metagenomics and directed evolution based on recent bioinformatics driven approaches.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
DA would like to acknowledge the UGC Rajiv Gandhi National fellowship, New Delhi. The authors wish to acknowledge the Head, Department of Biotechnology, D.D.U. Gorakhpur University, Gorakhpur for providing the infrastructural support.
References
- Kumar, R., Singh, S., Singh, O. Bioconversion of lignocellulosic biomass: biochemical and molecular perspectives. Ind. Microbiol. Biotechnol,2008; 35,374–379. doi: 10.1007/s10295-008-0327-8.
CrossRef - Juturu, V., Wu, J.C. Microbial xylanases: Engineering, production and industrial applications. Advanc, 2012; 30, 1219-1227.
- Paes, G., Berrin, J.G., Gabriel, J.B. GH11 xylanases: Structure/ function/properties relationships and applications. Advances, 2012;30, 564–592.
- Lafond, M., Guais, O., Maestracci ,M., Bonnin, E., Giardina, T. Four GH11 xylanases from the xylanolytic fungus Talaromycesversatilis act differently on (arabino) xylans. 2014; 98:6339–6352.
- Chakdar, H., Kumar, M., Pandiyan, K., Singh, A., Nanjappan, K., Kashyap, P.L., Srivastava, A.K. Bacterial xylanases: biology to biotechnology. 3 Biotech , 2016; 6:150.
CrossRef - Beg, Q.K., Kapoor, M., Mahajan, L., Hoondal, G.S. Microbial xylanases and their industrial applications: a review. ApplMicrobiolBiotechnol ,2001;56:326–338.
CrossRef - Collins, T., Gerday ,C., Feller, G . Xylanases, xylanase families and extremophilicxylanases. FEMS MicrobiolRev , 2005;29:3–23.
CrossRef - Nair, S.G., Sindhu, R., Shashidhar, S. Fungal xylanase production under solid state and submerged fermentation conditions. Afr J MicrobiolRes , 2008;2:82–86.
- Juturu, V., Wu, J.C. Microbial exo-xylanases: a mini review. ApplBiochemBiotechnol , 2014;174:81–92.
CrossRef - Walia, A., Guleria, S., Mehta, P., Chauhan, A., Parkash, J. Microbial xylanases and their industrial application in pulp and paper biobleaching: a review. 3 Biotech, 2017; 7:11.doi 10.1007/s13205-016-0584-6.
- Shatalov, A.A., Pereira, H. Effect of xylanases on peroxide bleachability of eucalypt (E. globulus) kraft pulp. BiochemEng J, 2008; 40:19–26.
CrossRef - Valls, C., Vidal, T., Roncero, M.B. The role of xylanases and laccases on hexenuronic acid and lignin removal. ProcBiochem , 2010; 45:425–430.
- Singh, V., Pandey ,V.C., Agrawal, S. Potential of Laceyellasacchari strain B42 crude xylanase in biobleaching of kraft pulp. Afr J Biotechnol , 2013;12(6):570–579.
- Walia, A., Mehta, P., Guleria, S., Shirkot, C.K. Modification in the properties of paper by using cellulase-free xylanase produced from alkalophilicCellulosimicrobiumcellulans CKMX1 in biobleaching of wheat straw pulp. Can J Microbiol , 2015b; 61:1–11.
CrossRef - Arora, N., Banerjee, A.K., Mutyala, S., Murty, U.S. Comparative characterization of commercially important xylanase enzymes.Bioinformation, 2009; 3(10): 446-453.
CrossRef - Uzuner, U., Shi, W., Liu, L., Liu, S., Dai, S.Y., Yuan, J.S. Enzyme structure dynamics of xylanase I from Trichodermalongibrachiatum, BMC Bioinformatics,2010;11(Suppl 6):S12.
CrossRef - Mathur, N., Goswami, G.K, Pathak, A.N. In silico study of Bacillus brevisxylanase—structure prediction and comparative analysis with other bacterial and fungal xylanase. Biomed Data Min, 2015; 4:112. doi:10.4172/2090-4924.1000112.
CrossRef - Chang, H.X., Yendrek, C.R., Anolles, G.C., Hartman, G.L. Genomic characterization of plant cell wall degrading enzymes and in silico analysis of xylanses and polygalacturonases of Fusariumvirguliforme. BMC Microbiology, 2016; 16:147.doi 10.1186/s12866-016-0761-0.
CrossRef - Deshmukh, R.A., Jagtap, S., Mandal, M.K., Mandal, S.K. Purification, biochemical characterization and structural modelling of alkali-stable β-1,4-xylan xylanohydrolase from Aspergillusfumigatus R1 isolated from soil. BMC Biotechnol., 2016; 4;16:11.
- Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M.R., Appel, R.D., Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server;
(In)John M. Walker (ed) : The Proteomics Protocols Handbook, Humana Press, 2005.pp. 571-607.
CrossRef - Larkin, M.A., Blackshields, G., Brown, N.P., Chenna, R., McGettigan, P.A., McWilliam, H., Valentin, F., Wallace, I.M., Wilm, A., Lopez, R., Thompson, J.D., Gibson, T.J., Higgins. DG. Clustal W and Clustal X version 2.0. Bioinformatics, 2007; 23, 2947-2948.
CrossRef - Kumar, S., Stecher, G., Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular biology and evolution, 2016; msw054.
CrossRef - Timothy, L., Bailey.,Bodén, M., Fabian, A., Buske., Frith, M., Grant ,C.E., Clementi, L., Ren, J., Li, W.W., Noble, W.S. MEME SUITE: tools for motif discovery and searching, Nucleic Acids Research, 37:W202-W208, 2009.
- Liu, L., Cheng, J., Chen, H., Li, X., Wang, S., Song, A., Wang, M., Wang, B., Shen, J . Directed evolution of a mesophilic fungal xylanase by fusion of a thermophilic bacterial carbohydrate binding module. Process Biochem, 2011;46:395–398.
CrossRef - Polizeli, M., Rizzatti, A., Monti, R., Terenzi, H., Jorge, J.A., Amorim, D. Xylanases from fungi: properties and industrial applications. ApplMicrobiolBiotechnol, 2005; 67:577–591.
CrossRef - Guruprasad, K., Reddy,B.V.B., Pandit M.W. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence.Protein Engineering,1990 ;4 no.2 pp.155-161.
CrossRef - Walia, A., Mehta, P., Guleria, S.,Chauhan,A.,Shirkot, C.K. Molecular cloning and sequencing of alkalophilicCellulosimicrobiumcellulansCKMX1 xylanase gene isolated from mushroom compost and characterization of the gene product. Arch. Biol. Technol,2015; 58(6),913-922.
CrossRef - Kyte, J., Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. Mol. Biol, 1982; 157(1), 105-32.
CrossRef - Ellouze, O.E., Loukil, S., Marzouki, M.N. Cloning and molecular characterization of a new fungal xylanase gene from Sclerotiniasclerotiorum S2. BMB reports,
- Sapag, A., Wouters, J., Lambert, C., de Ioannes, P., Eyzaguirre, J., Depiereux, E. The endoxylanases from family 11: computer analysis of protein sequences reveals important structural and phylogenetic relationships. J Biotechnol, 2002; 95: 109–131.
CrossRef - Torronen, A., Mach, R.L., Messner, R., Gonzalez, R., Kalkkinen, N., Harkki, A., Kubicek, C.P. The two major xylanases from Trichodermareesei: Characterization of both enzymes and genes. Nature publishing group, 1992.
- Cervantes, J.A., Godínez, G.D., Flores, Y.M., Gupta, V.K., Reyes, M.A.A. Phylogenetic analysis of β-xylanase SRXL1 of Sporisoriumreilianumand its relationship with families (GH10 and GH11) of Ascomycetes and Basidiomycetes.Scientific Reports, 2016; 6:24010. doi: 10.1038/srep24010.
CrossRef - Liao, H., Zheng, H., Li, S., Wei, Z., Mei, X., Ma, H., Shen, Q., Xu, Y. Functional diversity and properties of multiple xylanases from PenicilliumoxalicumGZ-2.Scientific Reports, 2015; 5:12631.doi: 10.1038/srep12631
CrossRef - Mohammed, A., Guda, C. Computational approaches for automated classification of enzyme sequences. J ProteBioinformat, 2011; 4,147-152.
- Damborsky, J., Brezovsky, J. Computational tools for designing and engineering enzymes. Current Opinion in Chemical Biology, 2014;19:8-16.
CrossRef - Suplatov, D., Voevodin, V., Svedas, V. Robust enzyme design : Bioinformatics tools for improved protein stability. J, 2015;10, 344-355.
- Yadav, P.K., Singh, V.K., Yadav, S., Yadav, K.D.S., Yadav, D.In silicoanalysis of pectin lyases and pectinases sequences. Biochemistry (Moscow), 2009; 74(9), 1049-1055.
CrossRef - Dubey, A.K., Yadav, S., Kumar, M., Singh, V.K., Sarangi, B.K., Yadav, D. In-silico characterization of pectatelyase protein sequences from different source organism. Enzy.Res, 2010.
- Yadav, V., Yadav, D., Yadav, K.D.S. In-silico analysis of α-L-rhamnosidase protein sequences from different source organisms. Onl.J.Bioinform, 2010;11(2),293-301.
- Malviya, N., Srivastava, M., Diwakar, S.K., Mishra, S.K. Insight to sequence information of polyphenol oxidase enzyme from different source organisms. Biochem.biotechnol, 2011; 165,397-405.
- Dubey, A.K., Yadav, S., Rajput, R., Anand, G., Yadav D. In silico characterization of bacterial, fungal and plant polygalacturonase protein sequences. Online Journal of Bioinformatics, 2012;13(2):246-259.
- Morya, V.K., Yadav, S., Kim, E.K., Yadav, D. In-silico characterization of alkaline proteases from different species of Aspergillus.Biochem.biotechnol, 2012;243-257.
- Yadav, S.K., Dubey, A.K., Yadav, S., Bisht, D., Darmwal, N.S., Yadav, D. Amino acid sequences based phylogenetic and motif assessment of lipases from different organisms. J.Bioinform, 2012; 13(3),400-417.
- Kumar, V., Singh, G., Verma, A.K., Agrawal, S. In Silico Characterization of Histidine Acid Phytase Sequences. Hindawi Publishing Corporation Enzyme Research, 2012; Vol, Article ID 845465, 8 pages.
- Dwivedi, V.D., Mishra, S.K .In silico analysis of L-asparaginase from different source organisms.Interdiscip Sci. 2014; 6(2):93-9.
CrossRef - Mathew, A., Verma, A., Gaur, S. An in-silico insight into characteristic of β-propeller phytase. Sci.Comput.Life Sci,2014; 6(2)133-139.
- Morya,V.K., Yadav, V.K., Yadav, S., Yadav, D. Active site characterization of proteases sequences from different species of Aspergillus.Biochem.Biophys,2016;74,327-355.
- Yadav, M., Yadav, S., Yadav, D., Yadav, K.D.S. In-silico Analysis of Manganese Peroxidases from Different Fungal Sources. Current Proteomics, 2017; 14, 201-203.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.



