

Manuscript accepted on : 14 March 2018
Published online on: --
Plagiarism Check: Yes
Genome Sequencing Analysis of Macrophomina Phaseolina Resistant and Susceptible Castor Genotype
Shulbhi Verma1, Rukam Singh Tomar2, Visha Rathode2, Jalpa Thakker2, Shubham2, Nawade Bhagwaat2, Sacheen Raval2, Tushar Antala2, Zeel Jogia2 and B. A. Golakiya2
1Department of Biotechnology, SradarkrusinagarAgricultural University, S.K Nagar.
2Department of Biotechnology, Junagadh Agricultural University, Junagadh.
Corresponding Author E-mail: itsshulbhi@gmail.com.
DOI : http://dx.doi.org/10.13005/bbra/2624

ABSTRACT: Castor (RicinusCommunis. L, 2n=20) is crop of tropical, sub tropical and warm temperate regions. Castor is most promising crop for the commercial and medicinal values. In recent years castor production is lessen due to Macrophomina Phaseolina diseases outbreak which is a necrotrophic soil borne pathogen. Macrophomina Phaseolina diseases known as root rot which cause severe diseases especially in dry area. In India it can damage 80-100% crop. No effective controlling measures are mentioned till date for disease. The more practical practices to control the disease are to identify castor resistant genotype from the Macrophomina Phaseolina fungus. There is very less information regarding the molecular aspect of castor JI 357 and 48-1 respectively resistant and susceptible genotype. Thus, Resistant genotype of castor provides basis of utilization of molecular approach in the molecular study of resistant genes and mechanism of their gene expression to increase the resistant genotype of castor whereas susceptible genotype helps in the compare the expression and activity of gene. Genome sequencing of resistant and susceptible castor genotype was carried out in Ion Torrent (PGM), Next Generation Sequencer. The data obtained in which resistant sample posses total 2,651,111 reads with total 379,341,629 bases with the average read length of 143 bp with 8 lowest and 597 highest sequence length with 43% of GC content in resistant genotype and in susceptible total 124,860 reads were generated with total 16,681,708 bases with the average read length of 134 bp with 8 lowest and 572 highest sequence length with 54% of GC content in susceptible sample of castor. Overall, 401Mb data was generated for resistant and susceptible sample with total reads 2,853,224. Blast2GO tool used to annotate the function of genes according to homologous sequence in resistant castor genotype JI357 out of 100,000 sequences, 89000 sequences were used for GO annotation, 3900 was blasted without hit and 2500 was blast with hit. Among that Ricinus communis have highest hits in resistant genotype while in susceptible 48-1 genotype of castor root in total 18,500 sequence, 4250 sequence used in with GO slim annotation, 9500 sequence used in mapping, 100 sequence found with blast hits and 4500 sequence is with blast (without hits) blast Go analysis.
KEYWORDS: Castor; Fungus; Genome; Sequencing
Download this article as:| Copy the following to cite this article: Verma S, Tomar R. S, Rathode V, Thakker J, Shubham S, Bhagwaat N, Raval S, Antala T, Jogia Z, Golakiya B. A. Genome Sequencing Analysis of Macrophomina Phaseolina Resistant and Susceptible Castor Genotype. Biosci Biotech Res Asia 2018;15(1). |
| Copy the following to cite this URL: Verma S, Tomar R. S, Rathode V, Thakker J, Shubham S, Bhagwaat N, Raval S, Antala T, Jogia Z, Golakiya B. A. Genome Sequencing Analysis of Macrophomina Phaseolina Resistant and Susceptible Castor Genotype. Biosci Biotech Res Asia 2018;15(1). Available from: https://www.biotech-asia.org/?p=29447 |
Introduction
Castor (Ricinus Communis L.) is a major kharif cash crop belongs to member of the Euphorbiacea. Castor is important perennial crop of arid and semi arid regions, tropical, semi tropical and warm temperate region of the world Weiss, (2000), Santos et al., (2007) . Castor oil is non edible and has been almost entirely for pharmaceutical, medicinal,cosmetics and industrial application Damodaran et al., (2007). Commercially castor is in demand which add the value in economy. The major castor producing countries are India, China and Brazil, accounting for 90% of castor produced in the world. India is the largest producer of castor, contributing to almost 65% of the world share. This share of castor can be enhanced through production but castor production growth can be constraint due to biotic and abiotic stress. Biotic stress is increasing day by day and tremendously reduces the crop. It is major constraint in the production of crops. Among them root rot caused by Macrophomina Phaseolina is a one of the major biotic threat for castor cultivation and causes significant yield losses(Anonymous, 2006). Macrophomina Phaseolina is a soil borne; global devasting necrotrophic fungal pathogen which found to be causal agent involved in a wilting disease of castor bean and infects more than 500 plants host inciting disease in a wide range of host’s genome and favored by hot and dry area (Yang et al., 2003)
The fungal infection and disease development are favored by high temperature and drought (Gupta et al., 2012). An early stage symptom of Macrophomina Phaseolina infection is appears of reddish brown lesion on the stem just above the soil. Dropping of leaves and branches, shredding of bark signifies the death of plant. The rotten tissues of Macrophomina Phaseolina contain large number of black or dark brown, thick walled sclerotia, fruiting bodies.
The breeding of resistant cultivars is the most effective economic and environmentally favorable method for controlling diseases. Macrophomina Phaseolina resistant is broadly categorized as either all stages resistance (seedling resistance) or adult plant resistance. But Macrophomina Phaseolina rapidly evolves new races, cultivars with seedling resistance to diseases which usually became susceptible within a few years after being popularized. Over the past several years, the rapid next generation sequencing method has resulted in high throughput gene expression profiling, genome annotation and other discovery related to resistant gene and genome.
For deciphering plant pathogen interaction and genes involved in transcriptomics is a revolutionary functional genomics tool. Transcriptomics study will help in the identification of the genes related to the resistance for this disease which is the upmost priority for its control. The transcriptome contains entire set of RNA transcripts of a given cell for a specific developmental stage or physiological condition (Ozsolak et al., 2011). The transcript analysis (transcriptomics) is the most advanced strategies for this purpose. It identifies the genes which can control the important traits and give the vast view of plant pathogen interaction, which may provide the basis for progress in genetic improvement of the crop (Gupta et al., 2012). Most defense responses are accompanied by rapid transcriptional activation of many genes (>1% of the genome). The fundamental issue of any diseases is to globally and integratively understand the interactions between pathogens and their hosts by using fast and effective techniques (Durrant et al., 2000). This can vary with external environmental condition including the state of the pathogen and pathogenesis. Transcriptome data also elaborates the activity of genes that change their expression patterns in response to a signal originated from the host plant tissue and reveal the mechanism of pathogenesis as initiated by fungal pathogen (Wong et al., 2005). The RNA sequence that obtained from transcriptome is very useful to investigate differences in gene expression patterns between the different samples of the population (Wolf et al., 2010) and in transcriptional profiles in the organisms (Morozova et al., 2008) RNA-Seq is a part of transcriptome so its data can be efficiently used to improve gene model prediction and to identify novel transcripts (Wang et al., 2009, Li et al., 2011, Gan et al., 2011). So it is the most prominent tool to study and understand gene function mainly because of its throughput and extensiveness.
An increasing body of evidence suggests that transcription sequencing using NGS technology provides high resolution data and is a powerful tool for studying global transcriptional networks. Using this technology, we can successfully obtained a large amount of sequence data that may represents the gene expression profiling of castor under particular condition. To be best of our knowledge, the transcription profiling of the castor genotype of JI357 resistant and 48-1 susceptible has not yet been reported. Considering the agricultural value of castor, a comprehensive description of the genes expressed in castor during infection is necessary for the discovery of candidate defense related genes. In this manuscript, we present the current understanding of the assembled and annotated transcription sequences. Additionally, we provide information obtained from a digital gene expression system that was used to compare the gene expression profile of adult castor plants at infection stages with Macrophomina Phaseolina. This comparison allowed us to elucidate the molecular mechanism and identify the responsive genes underlying the complex castor resistance Macrophomina Phaseolina. These findings should facilitate the developing of effective strategies for the breeding of resistant castor varieties to obtain a better control of root rot.
Molecular study of castor gene activity upon the infection of root rot is very limited so improved variety of castor from Macrophomina Phaseolina pathogen is demandable for the crop improvement. Exploration of the genome sequencing of resistant and susceptible castor genotype will enhance the chance of castor resistant genotype development and compare the difference of gene expression (Xin et al., 2012). Till date, genetic improvement of castor was based on the mass selection and pedigree methods for developing elite genotype with desirable attributes. This research will open the path on molecular biology aspect for crop improvement and will provide valuable genomic resources for further understanding the molecular basis of resistance mechanism (Hulbert, 2001). So, development of castor variety which are the rich reservoir of genes for resistance against biotic stress and introgression of these genes may be an option for improving yields and also other important traits in cultivated crop. So, best technology is to find out, identify and analyze the resistance gene. Plant disease resistance genes (R genes) encode proteins that detect pathogens. This R gene-mediated resistance has several attractive features for disease control (McDowell et al., 2003). There is several resistance genes present in the plants in that some activated during pathogen interaction that can also utilized in the development. Till defense mechanism is not in limelight just because molecular study is not well understood. To study these types of genes transcriptomics will be valuable tool to understand the resistant mechanism.
Material and Method
Plant materials and mRNA Extraction
Castor (Riccinus communis) genotype JI357 resistant to Macrophomina Phaseolina and 48-1 susceptible was used in the study. The plants were grown in field of Pathology department of Junagadh Agricultural University, Junagadh, Gujarat. The samples were collected at the fully matured condition by the end of January. Root samples were collected from castor resistant and susceptible plant from the farm and stored at -4°C in RNA later solution until their use for total RNA extraction. Total RNA was isolated from the 250mg of root from sample using mir Vana miRNA isolation kit and mRNA from total RNA by using Dynabeads® mRNA DIRECTTM Micro kit. Quality and quantity of RNA and mRNA were checked on Qubit.
cDNA library Preparation and Sequencing
For the NGS analysis, fragment the mRNA by Ion total RNA seq-kitv2 to size the sequence of desired length and use purified mRNA to construct a cDNA using Ion total RNA seq kit v2(Life te4chnologies, USA). The diluted cDNA library was sequenced as per manufacturer’s instructions on Ion torrent Sequencer.
For genome sequencing, fragmentized the RNA enzymatically to construct a library for further use as template preparation. Genome sequencing was carried out using Ion -314 chip in Ion Torrent Personal Genome Machine (PGMTM) from life technologies, at Department of Biotechnology, Junagadh Agricultural University, Junagadh, India as per the manufacture’s guidelines.
Transcriptome Assembly
To obtained clean reads, raw reads including reads with adapters, reads in which unknown bases represented more than 5% of the total bases, and low quality reads (percentage of low quality bases with a quality value ≤5 in more than 50% of a read),were removed. The clean reads were then assembled into longer contigs based on overlapping regions using the trinity platform. Contigs from a different transcript and their distances were confirmed by mapping clean reads back to the corresponding contigs , and thus, the transcript were defined as unigenes.
Genome Annotation
Raw reads of the sequence were processed for the quality control through default plug-in in Ion Torrent software server FAST QC. The quality reads were assembled in CLC Genomics Workbench v8.5 (CLC bio, Denmark). Contigs were ordered and aligned with reference genome Arabidopsis thallina (Parchman et al., 2010).
Annotation and Classification of Unigenes
Unigenes were annotated using BLASTX against various databases, including nr (NCBI non redundant protein databases), Swiss-prot, Gene ontology(GO) , Kyoto Encyclopedia of genes and genome (KEGG), and clusters of Ortologous Group, using a cut off E value of 10-5. Assignment of unigenes to pathways performed by searching the KEGG databases. The coding sequences of unigens were determined based on orthologous protein. The analysis mapped all of the annoted unigenes to GO terms in the database and counted the number of unigenes associated with each term. GO functional classification for the unigenes with GO term hits to view the distribution of gene functions of the species at the macro level.
Mapping Reads to Reference Genome
RNA seq libraries quality checked by using Fast QC and Fast X toolkit. Fast X toolkit (FASTX-Toolkit, 2015) contains FASTA or FASTAQ files which contain short quality reads sequence without , Denmark) to carry information of average quality sequence score (give data), read length distribution (data). Quality reads were obtained by trimming the raw reads at a minimum PHRED score of Q=20. The resulting high quality reads were mapped against reference genome viz., Arabidopsis thallina. Based on the percentage of reads mapped, further processing of Riccinus communis transcriptome data viz., annotation and pathway mapping was performed by using Arabidopsis as the reference genome (Ward 2012). Data was normalized by calculating the read per kilobase per million mapped reads (RPKM= Total exon reads/mapped reads in million X exon length in kb) for each gene and annotated using Blast2GO. Data of the transcriptome has been submitted to NCBI with data submission portal under SRA submission accession ID SRX1054812. Reads showing significant homology against Arabidopsis genes were mapped onto metabolic pathways using KEGG database and gene ontology. GO enrichment analysis provides all of the GO terms that are significantly enriched in differentially expressed genes compared with the genome background and filters the reads that correspond to biological functions. (Yingbin Hao et al., 2016)
The assembled sequences were compared against the NCBI non redundant protein database using BLASTX with cut-off E-value of 10-6. To annotate the assembled sequences according to gene ontology (GO Terms), the BLAST results were analyzed using Blast2Go to determine and compare gene functions. The GO terms were assigned to the representative transcripts for each sample through an enrichment analysis using Fisher’s exact test (p-value<0.01), with a false discovery rate correction in terms of biological processes, cellular processes and molecular functions. The transcript sequences were also aligned against Arabidopsis thallina using blastN in both alignments a cut off E value of 10-6 was applied. The BLAST search was limited to the first top significant query hits, and the gene names were assigned to each query based on the highest score.
Result and Discussion
Roots of resistant castor genotype was healthy inspite of Macrophomina phaseolina infection was introduced in the castor JI357 while susceptible genotype (48-1) could not resist itself from the disease. Roots of healthy castor genotype JI357 and disease castor genotype harvested by the end of January month almost after 100 days. This analysis starts with the RNA isolation followed by cDNA formation for library construction and used for sequencing in the Ion Torrent. Library was loaded in Ion 314 chip, in the run report 68% loading is done (Figure-1) and the library mean read length is 15.4bp. Raw reads of the sequencer were processed for the quality control through default plug-in in Ion Torrent software server(FAST QC). The quality reads were assembled in Genomics Workbench v8.5 (CLC bio, Denmark) which produced the data resulted in contigs and ordered with aligned with reference genome Arabidopsis thaliana using CLC genomics workbench. In resistant assembled reads were 2,651,111 and it covers 287.3 Mb whereas in susceptible assembled reads were 124,860 and it covers 113.7 Mb.
 |
Figure 1: Run report for whole transcriptome of castor
|
Before mapping , quality filtering of transcriptome sequencing data was done. In which low quality sequences (limit= 0.05) and ambiguous nucleotides were removed. The average percentage of quality filtered reads was 99.64% in resistant and 52.22% in susceptible data.
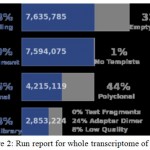 |
Figure 2: Run report for whole transcriptome of castor
|
 |
Figure 3: Read length resistant JI 357 and susceptible 48-1castor genotype before and after trim length.
|
RNA Seq Analysis of Resistant and Susceptible Castor Genotype
RNA seq provides the mapping with reference genome and quantification of gene. Sequencing of m RNA (RNA seq) can be used to quantify known genes, identify novel transcript and analysis within a sample (Mortazavi et al., 2008) it provides a wider range of expression level. To know the condition of response of castor plant upon Macrophomina Phaseolina infection, the transcriptome of infected plant was also sequenced using RNA seq approach and compared with the transcriptome from castor sample showing resistant towards infection. In resistant castor genotype JI357 matched and non matched reads with reference genome is 1,749,311 and 901,800 respectively whereas in susceptible castor genotype 48-1 this matched and non matched value is 44,739 and 80, 121 respectively (Table 1). In counted fragment unique transcript mapping number of reads in the mapping for the genes of uniquely assignable to the transcript were higher (245,802) in resistant genotype of castor JI 357 and lower (3,411) in susceptible genotype of castor after that these reads were assembled into 33968 and 2963 contigs respectively (Table 2). Contigs measurement at N50 (537) in castor resistant and (416) contigs measurement in castor susceptible genotype shown in (Table 3). The mapping was carried out by CLC software.
Table 1: Reads statistic of transcriptomic sequencing data of Resistant (JI357) and Susceptible (48-1) genotype
| Sample | Matched | Not matched |
| Resistant | 1,749,311 | 901,800 |
| Susceptible | 44,739 | 80,121 |
Table 2: Mapping statistics transcriptomic sequencing data of resistant (JI357) and susceptible 48-1 genotype
| Reads | % of Total Fragments | |||
| Resistant | Susceptible | Resistant | Susceptible | |
| Counted fragments | 295,881 | 5111 | 11.16 | 4.09 |
| – unique fragments | 245,802 | 3411 | 9.27 | 2.73 |
| – non-specifically | 50,079 | 1700 | 1.89 | 1.36 |
| Uncounted fragments | 2,355,230 | 119,749 | 88.84 | 95.91 |
| Total fragments | 2,651,111 | 124860 | 100.00 | 100.00 |
Table 3: Contigs measurements of transcriptomic sequencing data of Resistant (JI357) and Susceptible (48-1) genotype
| Sample | N50 | Contig |
| Resistant | 537 | 33,968 |
| Susceptible | 416 | 2,963 |
Differential Gene Expression in Castor Resistant and Susceptible Genotype Roots
Comparison factor of any two is important to reveal the difference as well as for measuring differential gene expression. For the comparison about the size of any group to another group is important, regarding the reference of fold change and t- statistic which are the two main choices for measuring differential expression because of relationship between reproducibility and accuracy, and between biological and statistical signal. In representation of differential gene expression, Volcano plot described to visualizes the display of both fold change and t-statistic. Fold change is the simplest method for identifying differentially expressed genes and is to evaluate the log ratio between two conditions mentioned as in the Figure 4. Differentially expressed gene of resistant and susceptible castor genotype in which some genes were upregulated genes and some were down regulated gene. Genes lies in the range of (p < 0.05) in the graph possess down regulated gene and genes lies in the range of (p > 0.05) shows upregulated gene. As we observe in terms of t test significant genes tend to locate in the upper left or upper right parts of the plot shown in Figure 4. Those genes are in the straight line shows no significant changes in the gene possible they are coexpressed with the change.
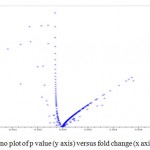 |
Figure 4: Volcano plot of p value (y axis) versus fold change (x axis)
|
So, Volcano plot of Kal’s test was illustrating the differentially expressed gene in JI357 resistant and susceptible 48-1 from castor root rot (Macrophomina phaseolina) and also represented up and down regulated genes in the doted volcano plot.
Identified differentially expressed gene by comparing the expression profile between infected and non infected castor root genotype. For each gene, the fold change in expression was calculated by dividing the normalized gene expression level in the infected plant by that in the corresponding uninfected. Select the gene which response and show significant differentially expressed gene by at least 2 fold in transcriptome resistant and susceptible genotype root. Based on their transcriptional levels in their relevant tissues, interaction of castor with Macrophomina phaseolina upregulated genes and its downregulated genes were identified. Overall approximately 19647 genes were upregulated and approximately 213 genes were down regulated in response to Macrophomina phaseolina in castor and few genes do not shown any the significant fold change it shows they were not affected by biotic stress.
These identified upregulated 19647 and downregulated 213 genes is from 100 days planting. Selection of 100 days assumed to be best time to compare differences in gene expression, because that most changes in gene expression occur 100 days when infection was fully developed in the plant. Number of gene identified in the upregulation and downregulation gene were identified clearly overlaps of the biological process, molecular process and cellular component.
Annotation of Resistant and Susceptible Castor Genotype
Annotation assigns the functional to gene and its gene products. Blast GO is one of the functional annotation tools which is quite easy and efficient. It performs functional annotation for each sequence based on the information available for the homologous sequences retrieved by BLAST. Fully infected castor genotype plant shows highest similarity with Macrophomina Phaseolina indicate the situation of disease whereas in resistant castor genotype Ricinus Communis genes shows highest similarity . In data distribution of bar chart of resistant JI357 genotype of castor root in total 100,000 sequences, 89000 sequences were used for GO annotation, 3900 was blasted without hit and 2500 was blast with hit was blast2go with annoted bar chart (Figure 5). While in susceptible 48-1 genotype of castor root in total 18,500 sequence, 4250 sequence used in with GO slim annotation, 9500 sequence used in mapping, 100 sequence found with blast hits and 4500 sequence is with blast (without hits) blast Go analysis Figure 5 (B).
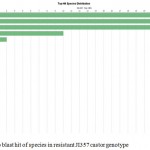 |
Figure 5a: Top blast hit of species in resistant JI357 castor genotype
|
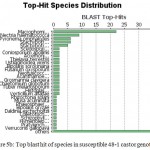 |
Figure 5b: Top blast hit of species in susceptible 48-1 castor genotype.
|
Gene ontology is distributed into three ontologies molecular function (MF), cellular component (CC), and Biological process (BP). The sequences could be categorized into 20 level two functional groups , which comprised three domain : biological process, cellular component and molecular function. Biological Process recognize series of events like cell development, metabolism, signal transduction. In the term of GO analysis of biological process it resulted in the identification of genes related to sepal, petal formation, ovule development and root hair elongation and the several protein involved in the biosynthetic process, small molecule metabolic process, cellular nitrogen compound metabolic process, response to stress and catabolic process were highly enriched in resistant castor genotype JI 357. While in susceptible 48-1 castor genotype biological process lowers in carbohydrate, cellular nitrogen, small molecule metabolic process, growth, response to stress and immune system. Molecular function defines ability of gene’s product like binding, enzyme, ligand etc. In GO analysis molecular function identified transcript for proton antiporter, cation antiporter activity, oxidoreducatase activity, kinase activity, transferase activity, hydrolase activity in highly enriched state. In susceptible molecular function ion binding ,oxidoreductase activity, ATP ase activity, RNA binding , enzyme binding, transmembrane transporter activity, structural constitute of ribosomes, DNA binding, hydrolase activity were enriched and its third component, cellular process recognize localization of cellular component within a cell like membrane, nucleus, complex (Hill et al., 2008). In GO term analysis cellular component category, protein involved with plastid, cytocol, nucleus, plasma membrane, mitochondira which were highly enriched. In susceptible cellular component protein involves mainly to cytocol, plasma membrane, extracellular region, mitochondria, nucleus, Golgi complex and several other organelle formation here plastid related protein is lower than resistant likewise many organelles effected due to disease incidence. To identify possible biological pathways that govern the responses of differentially expressed genes to the fungal treatment, GO category enrichment analysis was done using CLC Genomics.
 |
Figure 6: Data distribution of (A) resistant JI357 (B) susceptible 48-1 castor root sample bar chart
|
In Mapping database sources of resistant root JI357 castor genotype, in that maximum number of total GOs 5700000 found in UNIPROTKB data base while in susceptible root 48-1 castor genotype, maximum number of total GOs 1350000 found in UNIPROTKB data base, so comparatively less number of total GOs found in susceptible root sample.
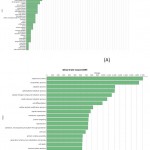 |
Figure 7: Direct GO count of biological process of (A) resistant JI357 and susceptible 48-1 castor genotype.
|
 |
Figure 8: Direct GO count of cellular component of (A) resistant JI357 and (B) susceptible 48-1 castor genotype.
|
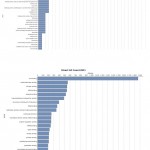 |
Figure 9: Direct GO count of molecular process of (A) resistant JI357 and (B) susceptible 48-1 castor genotype.
|
The different expression levels of up-regulated and down-regulated in different subcellular locations in the cytosol and the cell wall is consistent with the role of extracellular and intracellular components in activating gene expression in the response to Macrophomina phaseolina attack. The up-regulated and down-regulated encode proteins with roles in signal transduction pathways and resistance to Macrophomina phaseolina. The data obtained from our analysis indicate that there were more upregulated genes as compared to down regulated, implying that even in a state of stress the organism is actively adjusting its transcriptome and more, specifically. These findings suggest that a unique program of gene expression is activated in response to Macrophomina phaseolina. Number of responsive genes to Macrophomina phaseolina infection identified, was part of defence network in various plant pathogen interaction and disease resistance gene. Genes which was involved in the biosynthesis pathways of signal transduction and in plant pathogen interaction of KEGG (Kidd et al., 2011). There is large number of genes were expressed in infections and non infections castor plant but it is quite difficult to summarize each gene here. Therefore, on the basis of RPKM value and their fold change within the castor resistant and susceptible root samples, first top genes of up and down regulated during biotic stress were analyzed.
Table 4 : Functional gene found in Resistant castor JI357
| S.No | GO number | Type | Protein with locus | Fold change |
| 1. | GO:0000045 | M. P | transmembrane protein 93 | 7 |
| 2. | GO:0004674 | M. P | receptor-like serine threonine-protein kinase | 106 |
| 3. | GO:0003677 | M. P | histone h1 | 5 |
| 4. | GO:0000822 | M. P | protein auxin signaling | 72 |
| 5. | GO:0005739 | C.C | aquaporin | 36 |
| 6. | GO:0005886 | C.C | sulfate transporter | 13 |
| 7. | GO:0005618 | C.C | dead-box atp-dependent rna | 3 |
| 8. | GO:0005618 | C.C | glycine-rich rna-binding protein | 1 |
| 9. | GO:0005576 | C.C | cytochrome p450 | 1 |
| 10. | GO:0071555 | B.P | homeobox-leucine zipper protein athb-14 | 80 |
| 11. | GO:0006096 | B.P | adenosylhomocysteinase | 332 |
| 12. | GO:0009644 | B.P | ferredoxin-dependent glutamate synthase | 54 |
| 13. | GO:0000271 | B.P | protein auxin signaling | 72 |
| 14. | GO:0000209 | B.P | ubiquitin-protein ligase | 18 |
| 15. | GO:0006767 | B.P | glutamine-dependent NAD(+) synthetase | 136 |
| 16. | GO:0009698 | B.P | basic helix-loop-helix protein 147 | 1 |
Note: M.P (Molecular Function), C.C (Cellular Component), B.P (Biological Process).
Table 5: Functional gene found in Suceptible castor 48-1
| S.No | GO number | Type | Protein with locus | Fold change |
| 1. | GO:0016491 | M.F | Catalase ( EKG13864.1) | 78 |
| 2. | GO:0016491 | M.F | Short-chain dehydrogenase/reductase SDR ( EKG21920.1) | 1 |
| 3. | GO:0016491 | M.F | FAD linked oxidase (EKG16174.1) | 1 |
| 4. | GO:0016740 | M.F | Glycosyl transferase family 1 ((EKG16141.1) | 4 |
| 5. | GO:0003735 | M.F | structural constituent of ribosome ( EKG12572.1) | 4 |
| 6. | GO:0016787 | M.F | Glycoside hydrolase family 61 ( EKG21093.1) | 2 |
| 7. | GO:0004553 | M.F | Glycoside hydrolase family 7( hydrolyzing O-glycosyl compounds) ( EKG18618.1) | 7 |
| 8. | GO:0004497 | M.F | Monooxygenase FAD-binding protein
( EKG18636.1) |
1 |
| 9. | GO:0030529 | C.C | ribonucleoprotein complex
( EKG12572.1) |
2 |
| 10. | GO:0005840 | C.C | Ribosome ( EKG12572.1) | 4 |
| 11. | GO:0005622 | C.C | Intracellular ( EKG12572.1) | 2 |
| 12. | GO:0055114 | B.P | oxidation-reduction process FAD/NAD(P)-binding protein
(EKG15160.1) |
3 |
| 13. | GO:0008152 | B.P | metabolic process gene (EKG21920.1) |
3 |
| 14. | GO:0055114 | B.P | Monooxygenase FAD-binding protein
(EKG18636.1) |
2 |
| 15. | GO:0006950 | B.P | response to stress ( EKG17948.1) | 23 |
| 16. | GO:0006412 | B.P | Translation (EKG12572.1) | 6 |
| 17. | GO:0000272 | B.P | polysaccharide catabolic process
(EKG18618.1) |
4 |
| 18. | GO:0055114 | B.P | FAD linked oxidase (EKG16174.1) | 1 |
KEGG Pathways in Castor Genotype
There were several KEGG pathways found in resistant and susceptible castor root genotype. Among that most appropriate pathways considered in the research. In KEGG pathways of castor plant hormone and their signal plays important role in development and growth. In the pathway auxin hormone related to tryptophan metabolism which help in cell enlargement and growth, Zeatin biosynthesis related of cytokinine helps cell division and shoot initiation. In macrophomina phaseolina infected plant, enzymes of glycolosis is found which help in the formation of terpenoid backbone biosynthesis metabolism (precursor of zeatin). In the list of hormones there is another related hormone pathways are cysteine and methionine, barssinosteroid biosynthesis, α linolenic acid, Phenylaline metabolism. In plants, cysteine biosynthesis plays a central role in fixing inorganic sulfur from the environment and provides the only metabolic sulfide donor for the generation of methionine, glutathione, phytochelatins, iron-sulfur clusters, vitamin cofactors, and multiple secondary metabolites. Two enzymes catalyze the formation of cysteine in plant Serine acetyltransferase transfers acetate from acetyl-CoA to serine, generating O-acetylserine. O-Acetylserine sulfhydrylase (OASS or O-acetylserine (thiol)lyase) uses pyridoxal 5_- phosphate (PLP) as a cofactor to catalyze the formation of cysteine from O-acetylserine and sulfide. O-Acetylserine sulfhydrylase (OASS) catalyzes the final step of cysteine biosynthesis, the pyridoxal 5_-phosphate (PLP)-dependent conversion of O-acetylserine into cysteine. Although OASS is constitutively expressed, sulfur, nitrogen, and carbon starvation conditions increases cysteine biosynthesis in response to sulfur stress, and provides protection against oxidative stress (Bonner et al., 2005). In The sulphur-containing amino acid Met is essential in all organisms as a building block of proteins and as a component of the universal activated methyl donor S-adenosylmethionine (AdoMet). The synthesis of AdoMet accounts for 80% of Met metabolism, whereas the synthesis of proteins (the only pathway consuming the entire Met molecule) drives 20% of Met (Ravanel et al., 2004). The importance of carotenoids for plant growth and development is by major phytohormones abscisic acid, are derived from carotenoid precursors. Carotenoids are lipophilic secondary metabolites derived from the isoprenoid pathway and are accumulated in most plant organs. Carotenoids can exert important physiological functions in a wide range of organisms, including plants. They are essential components of the photosynthetic machinery, and play a critical role in preventing photo oxidative damage.
 |
Figure 10: Combined GO level distribution of (A) resistant JI357 (B) susceptible 48-1 castor genotype.
|
Abscisic acid accumulation has strong relation with plant–pathogen interactions it was shown in the susceptibility of rice plants towards Magnaporthe grisea. Abscisic acid is a key factor in the suppression of disease resistance to M. grisea. With regard to Abscisic acid induced susceptibility, it is worth noting that several fungal pathogens can produce Abscisic acid to infect the plant. The signaling responses of plants to ABA and biotic stress share many similarities that might act as additional nodes of competitive or synergistic interaction. The rapid generation of reactive oxygen species (ROS) is a central component of disease resistance responses and of ABA signalling (Flors et al., 2005). Brassinosteroids (BRs) are steroid hormones that affect virtually every physiological process of plants throughout their life cycle. Brassinosteroids (BRs) are poly hydroxylated steroid hormones that regulate various aspects of plant growth and development, such as vascular system differentiation, cell division and elongation, and sex determination (Yuhee et al., 2013). In vascular plants, some of the physiologically important secondary metabolites derived from Phenylalanine include pigments and defense molecules (Cho et al., 2007). Involvement of fatty acids and their oxidized derivatives in plant–pathogen interactions. Free linolenic as potent activators of defense reactions. These fatty acids are likely to be released in during pathogen attack, either directly by action of the pathogen on the cell membrane or by phospholipase activation in the signal transduction chain following elicitor recognition. Derivative of linolenic acids were shown to induce plant defense genes (Prost et al., 2005). Like importantly, JA has been shown to be a key defense molecule that interacts with additional defence pathways to control resistance to necrotrophic pathogens. All these hormone involve in different metabolism cause the same effect in the above mentioned pathway. This flow chart is of plant hormone and signal transduction pathway in castor plant (rcu04075).
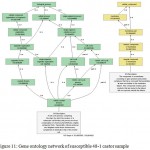 |
Figure 11: Gene ontology network of susceptible 48-1 castor sample
|
KEGG pathways for Plant Pathogen Interaction
Macrophomina phaseolina is the necrotrophs type of fungus. These necrotroph fungus often enter the plant through wounds and cause immediate and severe symptoms. Pathogen colonies inside the plant tissue by secreting toxins and degrade the cell wall, allow their hyphae to penetrate the plant cell wall and the cell themselves. Macrophomina phaseolina create vascular type of infection in which it block the infected vascular vessels and cause wilting and discoloration. Theses necrotrophs have little effect on plant physiology, since they kill host cells before colonising them. There was several physiology change happens in host plant due to fungal infection as shown in the study. It effects the physiology of the plant as we see in the respiration rate of plant which is invariably increases after fungi infection. It affects the rate of glucose catabolism causes a measureable increase in the temperature of infected parts. An early step in the plant’s response to infection is an oxidative burst is involved in a range of disease resistance and wound repair mechanism to rapid active defence. In resistance plant, the increase in respiration and glucose catabolism is used to produce defence related metabolites via pentose phosphate pathways.
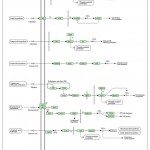 |
Figure 12: KEGG Pathway of Plant Hormone and Signal Transduction.
|
In susceptible plants, the extra energy produce is used by the growing pathogen. Pathogen also effects photosynthesis both directly and indirectly they decrease the photosynthetic rate by damaging chloroplast and killing the cells. This effects the translocation within the plant they depleted, diverse and retention of photosynthetic product by pathogen stunts plant growth, and further reduce the plants photosynthetic efficiency. Macrophomina phaseolina infect the root system directly which affects the plant’s ability to absorb water by killing the root system tissue, thus producing secondary symptoms of wilting and defoliation. Pathogen of vascular system affects water movement by blocking xylem vessels. Pathogen attack alters hormonal balance in plant by either by releasing plant hormone themselves, or by triggering an increase or a decrease in synthesis or degradation of hormone in the plant. These are the Physiological change occur in plant response against the pat this phenomenon it is clear that plants evolved a specific system with multiple layers against invading pathogens. The primary response includes the perception of pathogens by cell-surface pattern-recognition receptors (PRRs) and is referred to as PAMP-triggered immunity (PTI). Activation of FLS2 and EFR triggers MAPK signaling pathway that activates defense genes for antimicrobial compounds as shown in KEGG pathway of Figure 14 after the infection with Macrophomina phaseolina. As we know every plant show the pathogen attack so by this phenomenon it is clear that plants evolved a specific system with multiple layers against invading pathogens. The primary response includes the perception of pathogens by cell-surface pattern-recognition receptors (PRRs) and is referred to as PAMP-triggered immunity (PTI). Activation of FLS2 and EFR triggers MAPK signaling pathway that activates defense genes for antimicrobial compounds as shown in KEGG pathway of Figure 13
 |
Figure 13: KEGG Pathway of Plant Pathogen Interaction.
|
The increase in the cytosolic Ca2+ concentration is also a regulator for production of reactive oxygen species and localized programmed cell death/hypersensitive response, the secondary response is called effectors-triggered immunity (ETI). Pathogens can acquire the ability to suppress Pathogen triggered immunity (PTI) by directly injecting effector proteins into the plant cell through secretion systems. In addition, pathogens can manipulate plant hormone signalling pathways to evade host immune responses using coronatine toxin. Some plants possess specific intracellular surveillance proteins (R proteins) to monitor the presence of pathogen virulence proteins. This ETI occurs with localized programmed cell death to arrest pathogen growth, resulting in cultivar-specific disease resistance. Several elicitors of microbial origin have been identified as primary alarm signal molecules to switch on the plant immune systems culminating in activation of defence gene. PAMPS directly binds to plant pattern recognition receptors (PRR) and induced defence responses PAMPS trigger innate immune response in fungal cell wall derived chitin.
When the PAMP chitin synthesis was disrupted in the fungal pathogen, virulence of genes reduces the virulence of the pathogen. PAMP are conserved microbe specific molecules or signatures that activate the plant defence response in a manner analogous to the way in which molecules trigger an immune response. PAMP are often structural components of the pathogen cell wall or other conserved macromolecules. PAMP are perceived by PRR, which are divided into receptors like kinase proteins (RLK) and receptor like proteins (RLP). RLK have a cytoplasmic kinase domain that participates in intracellular signal transduction and an extracellular domain that is potentially responsible for ligand perception. RLP have structures similar to RLK but lack the cytoplasmic kinase domain. Damage associated molecular patterns triggered immunity here DAMPs are endogenous molecule with elicitor activity and are released from host cellular components during pathogen attack. Well characterized DAMP includes oligogalacturnoides, peptides, and cutin monomers. The response triggered by DAMP largely overlap with those activated by PAMP. Oligogalacturonides are derived from the degradation of major components of pectin in plant cell wall by microbial polygalacturonases during infections or by the action of endogenous polygalacturinases that are induced by mechanical damage. OGs elicit a wide range of defense responses, including an oxidative burst, accumulation of phytoalexins, an increase of glucanase, and chitinase activity, deposition of callose, increased hormone biosynthesis, and enhanced resistance to pathogen. Cell wall-associated receptor kinases have been considered potential receptors of OGs because of their ability to bind to OGs in vitro. WAK1, a member of the wall-associated kinase family, acts as a receptor of OGs as revealed by a receptor domain-exchange approach.
On the other hand, after the treatment with the cognate ligand elf1, the e EF-Tu receptor (EFR) ectodomain activates the WAK1 kinase that triggers defense responses similar with those activated by OGs and effective against fungal pathogens. In PAMP-PPR complex, several receptors for the PAMP have been recognized in plasma membrane of the plant cells. The PRR are modular protein harbouring an extracellular domain consists of leucine rich repeats or lysine motif. Most of the PRR are receptors like kinases (RLK) and the sensors for extracellular molecules consisting of an extracellular ligand binding domain, a single transmembrane domain, and a cytosolic protein kinases domain are called RLK. The extracellular domains of RLK bind directly to legands to perceive extracellular signals (PAMP), whereas the cytoplasmic kinases domins transducer these signals into the cell. Several secondary messengers encode information generated by the PAMP-PRR signalling complex and deliver the information of PRR to protein which interpret signals and initiates defence gene expression. Guanosine triphosphate (GTP) binding protein is the regulatory GTPase, which act as molecular switches in signal transduction system.
Two classes of signalling G protein, heteromeric G protein and small monomeric G protein (Ras/Ras like small GTPase). In the Ras superfamily of small GTPase, only the Ras and Rho families transmit extracellular signals. Ca++ is a master regulator of gene expression in plants calcium ion acts as a signal carrier. Calcium signalling is modulated by specific calcium signatures. Calcium ion is generated in the cytocol, and in noncytosolic locations including the nucleus and chloroplasts through the coordinate action of calcium ion influx and efflux pathways. Mitogen activating protein kinases cascades are major pathways that transducer extracellular stimuli into intracellular responses in plants. The oxidative burst involving rapid and transient production of reactive oxygen species (ROS) is a very rapid response, occurring within a second of PAMP treatment. ROS is a messenger in transmitting the PAMP signal .Nitric oxide has been identified as a gaseous second messenger. NO is a diffusible molecular messenger that play an important role in the plant immune response signal transduction system. PAMP triggers NO burst within minutes in plant cell. Plant hormone signals in plant immune signalling system in this section salicylic acid, ethylene, jasmonate, abscisic acid, cytokinin, gibberellins, and brassinosteriod plays important role in defence signalling against various pathogens. Jasmonic acid mediated pathways effectively confer resistance against necrotrophic fungal pathogen. Some of the PAMP may be contains within effectors. The innate immunity response is highly diverse in its capacity to recognize and respond to nectotrops, and most plants are resistant to most plant pathogen (Dodds et al., 2010).
The fungal effectors ethylene inducing xylanase (EIX) is an important factor for the success of pathogen invasion. EIX is a fungal effectors that contains a PAMP that is recognized by PAMP receptors. In EIX many types are there LeEIX 1 and LeEIX 2 which contain a leucine zipper, an extracellular LRR domain, a transmembrane domain, and a C terminal domain with an endocytosis signal. These PRR (LeEIX 1, LeEIX 2) are cell surface receptors without kinase domain in plants. The structure of these EIX receptors is similar to a family of receptor like proteins (RLP). These EIX receptors belongs to a superclade of leucine rich repeats receptors like protein with a signal for receptor mediated endocytosis, which was shown to be essential for proper induction of defence responses. The fungal effector ‘Avr’ from the pathogen contains a PAMP that interacts directly with the LRR of the R protein. Polygalacturonases and cellulose are produced by a wide range of pathogen and they acts as effectors and also function as general elicitors. During host pathogen interaction, many pathogens secrete these cell wall degrading enzymes.
These degrading enzymes can themselves function as elicitors, but their enzymatic products are also known to be general elicitors of plant defence responses .these enzymes degrade the plant cell wall structure and some of the degradation products such as pectin derived oligogalacturonides and cello dextrin’s act as potent elicitors of immunity. These host derived elicitors function almost in the same fashion as the PAMP function in plant innate immunity (Xuli et al., 2014). There is flow chart of plant pathogen interaction pathway in castor plant (rcu 04626).
Table 6: Gene related to Plant hormone and signal transduction KEGG pathway and Plant-Pathogen interaction KEGG pathway.
| S.No | Accession no | Gene | Fold change |
| 1. | RCOM1461650 | Chitinase 1 | 133 fold |
| 2. | RCOM0475040 | superoxide dismutase [cu-zn], putative | 128 fold |
| 3. | RCOM1620250 | serine/threonine protein kinase | 106 fold |
| 4. | RCOM0873840 | Catalase | 78 fold |
| 5. | RCOM0699100 | glutathione s-transferase, putative | 43 fold |
| 6. | RCOM1689300 | respiratory burst oxidase, putative | 18 fold |
| 7. | RCOM0191940 | Ethylene-responsive transcription factor 1A, putative | 14 fold |
| 8. | RCOM1409840 | Brassinosteroid Insensitive 1associated receptor kinase 1 precursor | 13 fold |
| 9. | RCOM0895260 | Disease resistance protein RPS5 | 12 fold |
| 10. | RCOM0723010 | big map kinase/bmk, putative | 5 fold |
| 11. | RCOM0654950 | Dead box ATP dependent RNA helicase | 3fold |
| 12. | RCOM1225970 | WRKY transcription factor, putative | 2 fold |
| 13. | RCOM_1500640 | receptor protein kinase | 2 fold |
| 14. | RCOM1160850 | Regulatory protein NPR1, putative | 1fold |
| 15. | RCOM_0645130 | leucine-rich repeat transmembrane protein kinase, putative | 1 fold |
NCBI Sequence Submission with Accession Number
The data of transcriptome have been submitted in National Centre for Biotechnology Information (NCBI) with Data submission portal under Sequence Read Archive (SRA) of resistant and susceptible castor genotype root sample and SRA Submission Accession ID: SRX1054812 with study summary linked to SRA is SRP059309 and Bioproject registered under accession: PRJNA286179. All the contigs were submitted to the gene bank and NCBI has published sequences data in July2015.
Conclusion
Genome analysis of the castor resistant genotype JI357 and susceptible genotype from Macrophomina Phaseolina fungus gives knowledge about the genes that are expressed in fungal stress condition and gene which are responsible for the resistant towards fungal invasion. The identified genes, expressed in transcriptome of resistant can be used to identify various signaling pathways responsible for regulating stress response, metabolism and plant defence mechanism. Further these genes can be used to develop markers for breeding program to transfer responsive gene from the resistant genotype by using marker assisted backcrossing .Therefore, these genes will be critically use for the understanding of the molecular mechanism involved in resistant states.
References
- Bonner E. R, Cahoon R. E, Knapke S. M and Jez J. M. Molecular Basis of Cysteine Biosynthesis in Plants .The Journal of Biological Chemistry. 2005;280(46):38803–38813.
CrossRef - Bioinformatics B. Fast QC.Cambridgeshire: Babraham Institute, 2015. Accessed. 2015 Nov 2. Available from http://www.bioinformatibabraham.ac.uk/projects/fastqc/.
- Cho M. H, Oliver R. A and Hong C .Y. Phenylalanine Biosynthesis in Arabidopsis thaliana. The Journal of Biological Chemistry. 2007;282(42):30827–30835.
CrossRef - Damodaran T and Hegde D. M. Oilseeds situation: a statistical compendium. Directorate of Oilseed Research, Hyderabad. 2007.
- Dodds P. N and Rathjen J.P. Plant Immunity: Towards an Integrated View of Plant Pathogen .Interactio Nature Reviews Genetics. 2010;11(8):539-548.
CrossRef - Durrant W. E, Rowl O, Piedras P, Hammond-kosack K. E, Lane C and Uh N. N. cDNA-AFLP reveals a striking overlap in race-specific resistance and wound response gene expression profiles. Plant Cell. 2000;12:963–977.
CrossRef - FASTX-Toolkit. Cold Spring Harbor: Cold Spring Harbor Laboratory, 2015. Accessed 2015 Nov 2. Available from: http://hannonlab.cshl.edu/fastx_toolkit/. 2015.
- Flors V, Ton J, Jakab G and Mani B. M. Abscisic acid and callose: team players in defense against pathogens? Phytopathol. 2005;153:1-7.
- Gan X, Stegle O, Behr J, Steffen J.G, Drewe P, Hildebrand K.L, Lyngsoe R, Schultheiss S.J, Osborne E.J, Sreedharan V.T, Kahles A, Bohnert R, Jean G, Derwent P, Kersey P, Belfield E.J, Harberd N.P, Kemen E, Toomajian C, Kover .X, Clark R.M, Rätsch G and Mott R. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. 2011;477:419–423.
- Gupta G. K, Sharma S. K and Ramteke R. Biology, epidemiolgy and management of the pathogenic fungus Macrophomina phaseolina (Tassi) Goid with special reference to charcoal rot of soybean (Glycine max (L.) Merrill). Journal of Phytopathology. 2012;160:167-180.
CrossRef - Hill D. P, Smith B, McAndrews M. S and Blake J. A. Gene Ontology annotations: what they mean and where they come from. BMC Bioinformatics. 2008;9(5):S2.
CrossRef - Hulbert S.H. Resistance gene complexes: evolution and utilization. Annu. Rev. Phytopathol. 2001;39:285–312.
CrossRef - Kidd N, KadooN. Y, Dombrecht B and Tekeoglu M. Auxin Signaling and Transport Promote Susceptibility to the Root-Infecting Fungal PathogenFusarium oxysporum in Arabidopsis. 2011;24(6):733-748.
- Li .Z, Zhang Z, Yan P, Huang S, Fei Z and Lin K. RNA-Seq improves annotation of protein coding genes in the cucumber genome. BMC Genomics. 2011;12:540.
CrossRef - Dowell J. M and Woffenden B. J. Plant disease resistance genes: recent insights and potential applications. Trends in Biotechnology. 2003;21(4):178-183.
CrossRef - Morozova O and MARRA M.A. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255-264.
CrossRef - Mortazavi A, Williams B. A, McCue K, Schaeffe L and Wold B. Mapping and quantifying mammalian transcriptomes by rna-seq. Nat Methods. 2008;5(7):621-628.
CrossRef - Ozsolak, and Milos P.M. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12:87-98.
CrossRef - Parchman T, Geist K, Grahnen J, Benkman C and Buerkle C. Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics11. 2010;180.
CrossRef - Prost I, Isabelle P, Sandrine D and Grit R. Evaluation of the antimicrobial activities of plant oxylipins supports their involvement in defense against pathogens. Plant Physiol. 2005;139:1902–1913.
CrossRef - Ravanel S, Block M. A, Rippert P, Jabrin S, Curien G, Rebeille F and Douce R. Methionine Metabolism in Plants. The Journal of Biological Chemistry. 2004;279(21):22548–22557.
CrossRef - Santos R. F, Kouri J, Barros M. A. L, Firmino P.T and Requião L. E.G. Aspectos Economics do Agronegócio da Mamoneira. In: O Agronegócio da Mamona no Brasil D.M.P. Azevedo & N.E. de M. Beltrão, (Eds.), Embrapa Informação Tecnológica, ISBN 978-85-7383-381-2, Brasília, Brazil. 22-41;2007.
- Wang Z, Gerstein M and Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev.Genet. 2009; 10:57–63.
CrossRef - Weiss E.A. Oilseed crops. 2nd Blackwell Science, Oxford. 2000.
- Wong M. L and Medrano J. F. Real-time PCR for mRNA quantitation. BioTechniques. 2005;39:75-85.
CrossRef - Wolf J.B,Bayer W.T and Haubold B. Nucleotide divergence versus gene expression differentiation: comparative transcriptome sequencing in natural isolates from the carrion crow and its hybrid zone with the hooded crow. Molecular Ecology. 2010;19:162–175.
CrossRef - Xin M, Wang X, Peng H, Yao Y and Xie C. Transcriptome Comparison of Susceptible and Resistant Wheat in Response to Powdery Milde Infection. Genomics Proteomics Bioinformatics. 2012;10:94–106.
CrossRef - Hao Y.W,Wang K, Wang X , Fu Y, Hung L and Kang Z. Trancriptome analysis provides insights into the mechanism underlying wheat plant resistance. PLoS ONE. 2016;11(3).
CrossRef - Yang X. B and Navi S. Charcoal Rot-A dry weather disease. Integrated Crop Management. 2003;22:166-167.
- Yuhee C and Sunghwa C. The Regulation of Brassinosteroid Biosynthesis in Arabidopsis. Critical Reviews in Plant Sciences. 2013;32:396–410.
CrossRef - Xuli W, Nan J, Jinling L and Wende L. The role of effectors and host immunity in plant–necrotrophic fungal interactions. Virulence. 2014;5(7):1–11.

This work is licensed under a Creative Commons Attribution 4.0 International License.



